
一种新型噁唑烷酮类抗生素的合成
2017-04-01 02:52赵胜贤胡红华李啸风任党培金晓鲁
合成化学 2017年3期
赵胜贤, 厉 昆, 胡红华, 李啸风, 任党培, 金晓鲁, 陈 治
(1. 浙江普洛得邦制药有限公司,浙江 东阳 322118; 2. 优胜美特制药有限公司,浙江 金华 321025)
·制药技术·
一种新型噁唑烷酮类抗生素的合成
赵胜贤1,2*, 厉 昆1, 胡红华2, 李啸风1, 任党培1, 金晓鲁2, 陈 治1
(1. 浙江普洛得邦制药有限公司,浙江 东阳 322118; 2. 优胜美特制药有限公司,浙江 金华 321025)
以2,4-二溴吡啶为原料,经Weinreb酰胺酰化、Noyori不对称氢转移反应、脱Boc保护基、环化、Suzuki偶联、磷酸单酯化及成盐共七步反应制得一种新型噁唑烷酮类抗生素【【(3R,3aS)-7-{6-[(S)3-甲基-2-噁唑烷酮-5-基]吡啶-3-基}-1-氧-1,3,3a,4-四氢苯并[b]噁唑[3,4-d][1,4]噁嗪-3-基】甲基】磷酸单酯二钠盐,其结构经1H NMR,13C NMR和HR-MS(ESI)确征,总收率7%,纯度99.7%。
2,4-二溴吡啶; 磷酸单酯; 噁唑烷酮; 抗生素; 药物合成
抗菌药物是临床上使用最频繁的药物之一,由于历史原因造成的抗生素不规范使用,导致细菌对现有抗生素严重的耐药性,多药耐药(MDR)的细菌感染已经成为全球公共卫生健康的主要威胁之一,世界正逐步进入“后抗生素时代”[1-3]。为扭转这一严峻局势,各国正加紧新型抗生素的研发,美国还出台了抗生素研发激励法案(GAIN法案)[4]。
噁唑烷酮类抗菌药物是一类二十一世纪新开发上市的具有新作用机制的全合成抗生素[5],如美国法玛西亚普强公司开发的利奈唑胺,该药于2000年经FDA批准在美国上市,是全球首个上市的噁唑烷酮类抗菌药物[6]。之后,美国Cubist公司开发的磷酸泰地唑胺也于2014年经FDA批准在美国上市[7]。
杨玉社课题组[8]开发了一种新型噁唑烷酮类抗菌药物【【(3R,3aS)-7-{6-[(S)3-甲基-2-噁唑烷酮-5-基]吡啶-3-基}-1-氧-1,3,3a,4-四氢苯并[b]噁唑[3,4-d][1,4]噁嗪-3-基】甲基】磷酸单酯二钠盐(1),其具有较同类药物更强的抗菌活性,尤其是抗多耐药菌活性。该研究以2,4-二溴吡啶(2)为原料,经氯乙酸的Weinreb酰胺酰化、Noyori不对称氢转移反应、叠氮亲核取代、胺化、环化、甲基化、Suzuki偶联、磷酸三酯化、氢化和成盐总共十步反应制得1,反应涉及叠氮钠和叠氮中间体易爆炸的试剂及中间体、a-氯代酮类易过敏的试剂和中间体、氢化等EHS隐患大的反应,而且a-氯代酮类化合物具有基因毒性,不适宜工业化药品生产[8-9]。
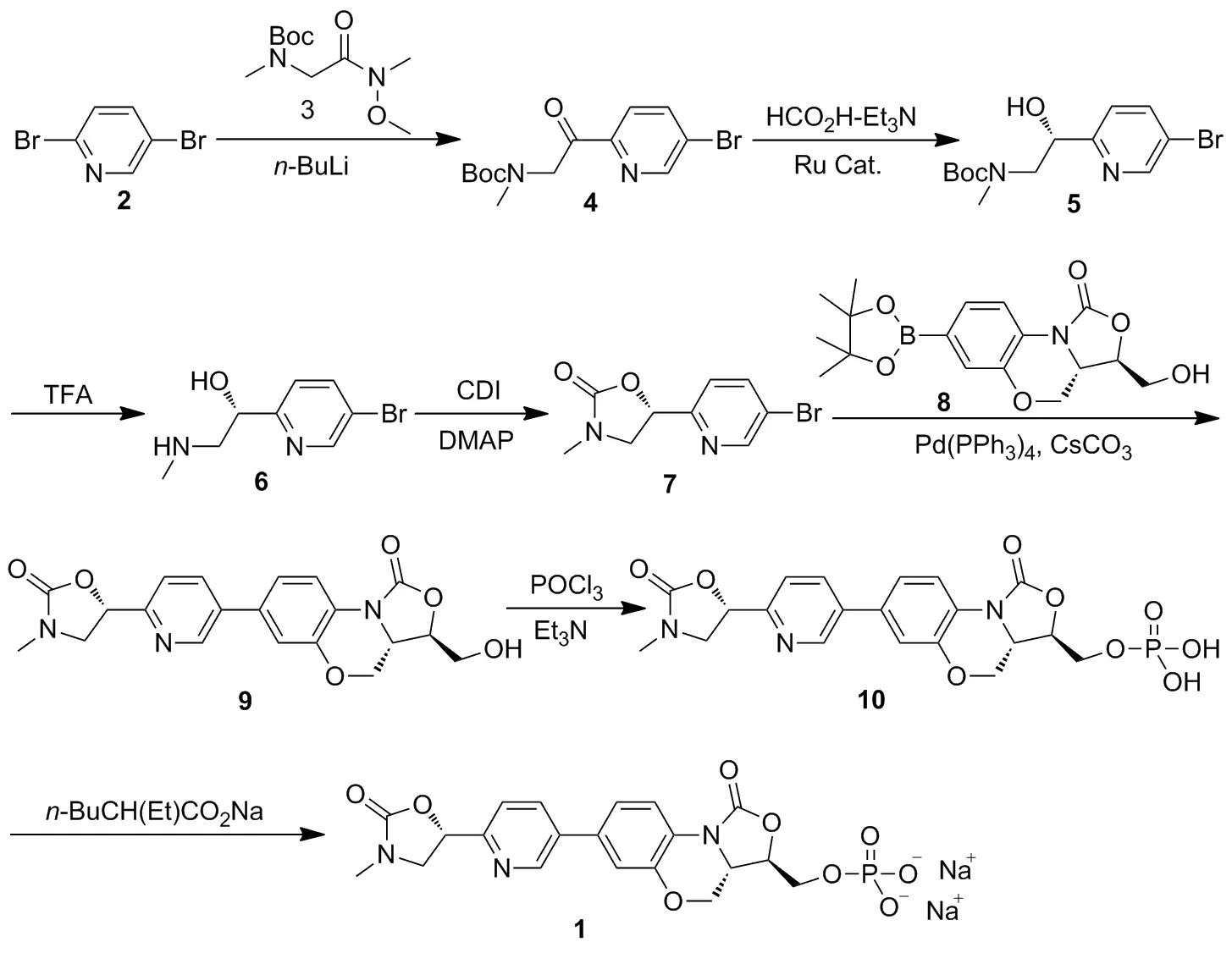
Scheme 1
本文在杨课题组研究的基础上进行了工艺改进,以2为原料,经Boc肌氨酸的Weinreb酰胺(3)酰化、Noyori不对称氢转移反应、脱Boc保护基、环化、Suzuki偶联、磷酸单酯化及成盐共七步反应制得1(Scheme 1),其结构经1H NMR、13C NMR和HR-MS(ESI)确征。该合成路线避免了叠氮化合物、a-氯代酮类化合物、氢化反应及柱层析纯化,具有反应路线短、工艺安全性高和产品纯度好等优点,适合工业化的生产。
1 实验部分
1.1 仪器与试剂
Brucker AvanceⅢ 500型核磁共振仪(CDCl3为溶剂,TMS为内标); AB Sciex Triple TOF 5600+型四极杆飞行时间质谱仪; Agilent 1260型高效液相色谱仪。
2,江苏鑫星科技有限公司;Boc肌氨酸的Weinreb酰胺,上海昊锐医药生物科技有限公司;(3R,3aS)-3-(羟甲基)-7-(4,4,5,5-四甲基-1,3,2-二氧杂硼烷-2-基)-3a,4-二氢苯并[b]噁唑[3,4-d][1,4]噁嗪-1(3H)-酮(8)参照文献[9]合成;其余所用试剂均为工业级。
1.2 合成
(1)[ 2-(5-溴吡啶-2-基)2-氧乙基](甲基)氨基甲酸叔丁酯(4)的合成
在反应瓶中加入2 23.69 g(0.1 mol)和甲苯100 mL,搅拌使其溶解,冷却至-78 ℃,滴加2.5 mol·L-1正丁基锂0.1 mol的正己烷溶液40 mL,滴毕,加入3 23.23 g(0.1 mol),于-78 ℃反应1 h。加入10%氯化铵溶液100 mL淬灭反应,分出甲苯层,依次用水洗涤,无水硫酸钠干燥,浓缩,经乙酸乙酯-正庚烷重结晶得白色固体4 16.83 g,收率51%,纯度98%(HPLC), m.p. 89.5~90.7 ℃;1H NMRδ: 1.37(s, 9H, rotamer 1), 1.49(s, 9H, rotamer 2), 2.96(s, 3H, rotamer 1), 2.97(s, 3H, rotamer 2), 4.80(s, 2H, rotamer 1), 4.88(s, 2H, rotamer 2), 7.91~8.02(m, 4H, rotamer 1 and 2), 8.71~8.72(m, 2H, rotamer 1 and 2);13C NMRδ: rotamer 1: 28.5, 35.9, 55.5, 80.0, 123.3, 125.9, 139.9, 150.4, 156.1, 195.7; rotamer 2: 28.6, 36.1, 55.8, 80.2, 123.4, 126.0, 140.1, 150.5, 156.6, 195.9; HR-MS(ESI)m/z: Calcd for C13H17N2O3BrNa{[M+Na]+}351.032 0, found 351.032 3。
(2) [2-(5-溴吡啶-2-基)2-羟乙基](甲基)氨基甲酸叔丁酯(5)的合成
在反应瓶中加入二氯(p-甲基异丙苯)钌(II)二聚体0.61 g(1 mmol), (1S,2S)-(+)-N-对甲苯磺酰基-1,2-二苯基乙二胺0.73 g(2 mmol),N,N-二甲基甲酰胺5 mL和三乙胺0.2 mL,于室温反应1 h;搅拌下加入甲酸-三乙胺共沸物43.25 g(摩尔比为5 ∶2)和甲基叔丁基醚100 mL,加毕,降温至0 ℃,加入4 32.92 g(0.1 mol),反应4 h。加入水100 mL搅拌5 min,分液,有机相经无水硫酸钠干燥,浓缩得黄色油状液体5粗品35.38 g,纯度97%(HPLC),ee86%,直接投入下一步反应;1H NMRδ: 1.38, 1.45(2s, 9H, rotamer 1 and 2), 2.82, 2.88 (2s, 3H, rotamer 1 and 2), 3.45~3.65(m, 2H), 4.88~4.95(m, 1H), 5.06(brs, 1H), 7.46~7.48(m, 1H), 7.81~7.83(m, 1H), 8.58~8.61(m, 1H);13C NMRδ: 28.5, 35.9, 36.7(rotamer 1 and 2), 56.2, 72.2, 73.9(rotamer 1 and 2), 80.0, 80.5(rotamer 1 and 2), 119.3, 119.6(rotamer 1 and 2), 122.4, 139.4, 149.6, 149.9(rotamer 1 and 2), 155.6, 158.1(rotamer 1 and 2), 159.2, 160.0(rotamer 1 and 2); HR-MS(ESI)m/z: Calcd for C13H20N2O3Br{[M+H]+}331.065 7, found 331.065 9。
(3)(S)-2-(N-甲基)氨基-1-(5-溴吡啶-2-基)乙醇(6)的合成
在反应瓶中加入粗品5 35.38 g,二氯甲烷100 mL和三氟乙酸50 mL,于室温反应8 h。反应液浓缩后加入二氯甲烷100 mL和水100 mL,用饱和氢氧化钠溶液调至pH 9.0,分液,有机层用水洗涤,浓缩,经乙酸乙酯-正庚烷重结晶得黄色固体6 14.76 g,两步合并收率64%,纯度99%(HPLC),ee95%, m.p.98.4~99.1 ℃;1H NMRδ: 2.46(s, 3H), 2.78(dd,J=12.0 Hz, 8.0 Hz, 1H), 2.99(dd,J=12.0 Hz, 4.0 Hz, 1H), 3.06 (br s, 2H), 4.80(dd,J=8.0 Hz, 4.0 Hz, 1H), 7.38(d,J=8.2 Hz, 1H), 7.82(dd,J=8.2 Hz, 2.2 Hz, 1H), 8.59(d,J=2.2 Hz, 1H);13C NMRδ: 36.4, 57.9, 71.4, 119.4, 122.0, 139.6, 149.9, 160.3; HR-MS(ESI)m/z: Calcd for C8H12N2OBr{[M+H]+}231.013 3, found 231.012 4。
(4) (S)-5-(5-溴吡啶-2-基)-3-甲基噁唑烷-2-酮(7)的合成
在反应瓶中加入5 23.11 g(0.1 mol),N,N′-羰基二咪唑(CDI)24.32 g(0.15 mol), 4-二甲氨基吡啶(DMAP)2.44 g(0.02 mol)和四氢呋喃100 mL,于室温反应3 h。反应液浓缩后加入二氯甲烷100 mL和1 mol·L-1盐酸100 mL,分液,有机层用水洗涤,浓缩,经乙酸乙酯-正庚烷重结晶得白色固体7 17.25 g,收率67%,纯度99%(HPLC),ee100%, m.p.72.1~72.4 ℃;1H NMRδ: 2.91(s, 3H, CH3), 3.71(dd,J=8.9 Hz, 6.2 Hz, 1H), 4.00(t,J=8.9 Hz, 1H), 5.51(dd,J=8.9 Hz, 6.2 Hz, 1H), 7.46(d,J=8.4 Hz, 1H), 7.89(dd,J=8.4 Hz, 2.2 Hz, 1H), 8.65(d,J=2.2 Hz, 1H);13C NMRδ: 31.2, 52.3, 73.4, 120.5, 121.8, 139.9, 150.8, 156.9, 157.8; HR-MS(ESI)m/z: Calcd for C9H10N2O2Br{[M+H]+}256.992 6, found 256.991 8。
(5) (3R,3aS)-3-(羟甲基)-7-{6-[(S)-3-甲基-2-噁唑烷-5-基]吡啶-3-基}-3a,4-二氢苯并[b]噁唑[3,4-d][1,4]噁嗪-1(3H)-酮(9)的合成
在反应瓶中加入7 25.71 g(0.1mol), 8 34.72 g( 0.1mol)和碳酸铯65.16 g( 0.2 mol), 加入二氧六环180 mL和水20 mL的混合溶剂搅拌使其溶解;加入Pd(PPh3)40.51 g,回流反应5 h。冷却至室温,抽滤,滤饼用水洗涤,经丙酮重结晶得灰白色固体9 29.43 g,收率74%,纯度99%,ee100%, m.p.>200 ℃;1H NMRδ: 2.82(s, 3H), 3.69~3.72(m, 2H), 3.77~3.80(m, 1H), 3.95~3.99(m, 1H), 4.08~4.10(m, 2H), 4.48~4.49(m, 1H), 4.59~4.60(m, 1H), 5.36(br s, 1H), 5.63(dd,J=9.0 Hz, 6.0 Hz, 1H), 7.40~7.42(m, 2H), 7.55(d,J=8.0 Hz, 1H), 7.99(d,J=8.7 Hz, 1H), 8.15(dd,J=8.7 Hz, 2.2 Hz, 1H), 8.93(d,J=2.2 Hz, 1H);13C NMRδ: 31.1, 51.7, 52.2, 61.3, 66.5, 73.8, 77.0, 115.5, 119.5, 120.3, 121.7, 124.2, 132.8, 135.0, 135.3, 145.2, 147.9, 154.2, 156.9, 157.7; HR-MS(ESI)m/z: Calcd for C20H20N3O6{[M+H]+}398.135 2, found 398.135 3。
(6) 【【(3R,3aS)-7-{6-[(S)3-甲基-2-噁唑烷酮-5-基]吡啶-3-基}-1-氧-1,3,3a,4-四氢苯并[b]噁唑[3,4-d][1,4] 噁嗪-3-基】甲基】磷酸单酯(10)的合成
在反应瓶中加入9 39.74 g(0.1 mol),二氯甲烷400 mL,三乙胺55.75 mL (0.4mol)和三氯氧磷46.00 g(0.3 mol),于室温反应过夜。加水200 mL,抽滤,滤饼经N,N-二甲基甲酰胺重结晶得淡黄色固体10 28.20 g,收率59%,纯度99%(HPLC,杂质包括6个非对映异构体),ee100%, m.p. 197.1~198.3 ℃;1H NMRδ: 2.82(s, 3H), 3.70~3.72(m, 1H), 3.97(t,J=8.8 Hz, 1H), 4.11~4.22(m, 4H), 4.64~4.69(m, 2H), 5.63 (dd,J=9.0 Hz, 6.0 Hz, 1H), 7.41~7.42(m, 2H), 7.55(d,J=8.4 Hz, 1H), 7.98(d,J=9.0 Hz, 1H), 8.15(dd,J=8.4 Hz, 2.2 Hz, 1H), 8.93(d,J=2.2 Hz, 1H);13C NMRδ: 31.2, 51.7, 52.0, 65.1, 66.3, 73.8, 74.8, 115.6, 119.7, 120.3, 121.8, 124.1, 133.0, 135.0, 135.4, 145.2, 147.9, 153.9, 156.9, 157.8; MS(ESI)m/z: 478.1{[M+H]+}。
(7) 1的合成
在反应瓶中加入10 4.77g(0.01 mol)、异辛酸钠6.65 g(0.04 mol)和水25 mL,搅拌使其溶解,溶清后滴加丙酮250 mL,滴毕,析出沉淀,抽滤,滤饼用丙酮洗涤,干燥得白色固体1 3.78 g,收率73%,纯度99.7%(HPLC,杂质包括6个非对映异构体),ee100%, m.p.>200 ℃;1H NMR (D2O, 500 MHz)δ: 2.75(s, 3H), 3.33~3.36(m, 1H), 3.79~3.81(m, 1H), 3.88(t,J=9.3 Hz, 1H), 3.96~4.04(m, 3H), 4.52~4.58(m, 2H), 5.31(dd,J=9.5 Hz, 7.0 Hz, 1H), 6.58(d,J=1.5 Hz, 1H), 6.70(dd,J=8.5 Hz, 1.5 Hz, 1H), 7.02(d,J=8.5 Hz, 1H), 7.37(dd,J=8.2 Hz, 1.9 Hz, 1H), 7.48(d,J=8.2 Hz, 1H), 8.16(d,J=1.9 Hz, 1H);13C NMRδ: 30.2, 52.0, 52.6, 63.6, 66.3, 74.1, 76.2, 114.5, 119.1, 119.4, 120.9, 122.7, 131.9, 134.4, 135.1, 144.2, 146.4, 154.8, 155.0, 159.4; HR-MS(ESI)m/z: Calcd for C20H21N3O9P{[Mfree acid+H]+}478.101 5, found 478.101 4。
2酰化的位置是吡啶环的2-位,而不是4-位。7的分子结构经(图1)确证。
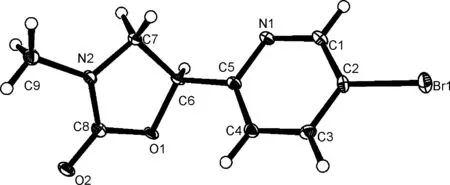
图1 7的分子结构图
4和5由于Boc取代基的空间位阻较大,导致单键旋转受阻,从1H NMR和13C NMR上可以发现,产物为一对旋转异构体(rotamer),其中4的两个旋转异构体的比例恰好为1 ∶1,文献报道3也存在相同的旋转异构体现象[10]。
反应采用3而不是氯乙酸的Weinreb酰胺为起始原料,有效避免了杨玉社课题组原工艺中涉及的叠氮钠和叠氮中间体易爆炸的试剂及中间体、α-氯代酮类易过敏的试剂和中间体[8-9],直接采用三氯氧磷合成磷酸单酯(10),也规避了原路线的氢化工序。工艺的安全性大大提高,更加适宜大规模的生产。由于a-氯代酮具有基因毒性,残留需要控制在百万分之一级别,因此本制备方法同时可以提高用药的安全性。
本合成路线的所有产品均通过重结晶提纯,革除了原工艺中的所有柱层析方法,适合产品的放大生产。本工艺的合成步骤由杨玉社课题组的原工艺的十步化学反应缩短到七步,总收率为7%,原工艺的总收率为9%[9]。虽然收率基本一致,但步骤的缩短有利于降低成本。另外,Noyori不对称氢转移反应步骤得到的中间体5未经过纯化,直接投入下一步反应,均有效降低了生产成本,工艺的经济性更加显著。
[1] Fukuda Y. New approaches to overcoming bacterial resistance[J].Drugs Future,2009,34(2):127-136.
[2] Spellberg B, Guidos R, Gilbert D,etal. The epidemic of antibioticresistant infections:A call to action for the medical community from the infectious diseases society of America[J].Clin Infect Dis,2008,46(2):155-164.
[3] Boucher H W, Talbot G H, Bradley J S,etal. No drugs:No ESKAPE!An update from the infectious diseases society of America[J]. Clin Infect Dis,2009,48(1):1-12.
[4] 张梦,邵蓉. 美国抗生素研发激励政策及启示[J].中国新药杂志,2016,25(1):13-18.
[5] 翟鑫,汪玉梅,宫平. 新型噁唑烷酮类抗菌药物的研究进展[J].沈阳药科大学学报,2006,23(11):739-744.
[6] 肖永红. Linezolid:第一个应用于临床的噁唑烷酮类抗菌药[J].国外医药抗生素分册,2001,22(6):279-281.
[7] 陈本川. 新型抗菌药:特地唑胺磷酸酯(tedizolid phosphate)[J].医药导报,2014,33(12):1679-1683.
[8] Yang Y S, Guo B. Benzoxazine oxazolidinine compound, preparation method and application thereof:EP 2940024A1[P].2015.
[9] Guo B, Fan H X, Xin Q S,etal. Solubility-driven optimization of (pyridin-3-yl) benzoxazinyl oxazolidinones leading to a promising antibacterial agent[J].J Med Chem,2013,56(6):2642-2650.
[10] Bhavesh H P, Andrew M M, Hetal P,etal. Conversion ofa-amino acids into bioactiveo-aminoalkyl resorcylates and related dihydroxyisoindolinones[J].J Org Chem,2011,76(15):6209-6217.
Synthesis of A Novel Oxazolidinone Antibiotic
ZHAO Sheng-xian1,2*, LI Kun1, HU Hong-hua2, LI Xiao-feng1, REN Dang-pei1, JIN Xiao-lu2, CHEN Zhi1
(1. Zhejiang Apeloa Tosopo Pharmaceutical Co., Ltd., Dongyang 322118, China; 2. Yosemade Pharmaceutical Co., Ltd., Jinhua 321025, China)
A novel oxazolidinone antibiotic, disodium 【【(3R,3aS)-7-{6-[(S)3-methyl-2-oxazolidino-5-yl]pyridin-3-yl}-1-oxo-1,3,3a,4-tetrahydrobenzo[b]oxazolo[3,4-d][1,4]oxazin-3-yl】methyl】phpsphate, was synthesized by seven-step reactions from 2,4-dibromopyridine,viaacylation by Weinreb amide, Noyori asymmetric transfer hydrogenation, removal of Boc protecting group, cyclization, Suzuki coupling, mono-phosphate esterfication and salification. The structure was confirmed by1H NMR,13C NMR and HR-MS(ESI). The overall yield and purity were 7% and 99.7%, respectively.
2,4-dibromopyridine; mono-phosphate; oxazolidinone; antibiotic; drug synthesis
2016-08-26;
2016-12-26
赵胜贤(1970-),男,汉族,浙江绍兴人,博士,高级工程师,主要从事药物化学及晶型的研究。 E-mail: 2086488660@qq.com
O626; R914.5
A
10.15952/j.cnki.cjsc.1005-1511.2017.03.16217
猜你喜欢
中国药学药品知识仓库(2022年10期)2022-05-29
安徽化工(2022年1期)2022-02-15
汕头大学学报(自然科学版)(2020年4期)2020-12-14
中华养生保健(2020年3期)2020-11-16
化工学报(2020年4期)2020-05-28
今日农业(2019年11期)2019-08-13
天然产物研究与开发(2018年7期)2018-08-21
天然产物研究与开发(2018年4期)2018-05-07
中成药(2017年5期)2017-06-13
中国塑料(2015年10期)2015-10-14
