
氟喹诺酮C-3噁二唑硫乙酰腙的合成及抗肿瘤活性测定*
2017-02-10 08:08张会丽胡国强黄文龙
郑州大学学报(医学版) 2017年1期
李 珂,张会丽,闻 婧,胡国强#,黄文龙
1)郑州工业应用技术学院药学院 郑州 451150 2)河南大学药学院 河南开封 475001 3)中国药科大学新药研究中心 南京 210009
氟喹诺酮C-3噁二唑硫乙酰腙的合成及抗肿瘤活性测定*
李 珂1),张会丽1),闻 婧2),胡国强2)#,黄文龙3)
1)郑州工业应用技术学院药学院 郑州 451150 2)河南大学药学院 河南开封 475001 3)中国药科大学新药研究中心 南京 210009
#通信作者,男,1964 年3 月生,博士,教授,研究方向:新药分子的设计与合成,E-mail:hgqxy@sina.com
氟喹诺酮;噁二唑;酰腙;生物电子等排体;抗肿瘤活性
目的:发现氟喹诺酮由抗菌活性转化为抗肿瘤活性的有效结构修饰策略。方法:用噁二唑杂环作为环丙沙星C-3羧基的生物电子等排体,硫乙酰腙为其功能修饰侧链,合成10个新氟喹诺酮C-3噁二唑硫乙酰腙目标化合物,其结构经元素分析和光谱数据确证,并评价其体外对SMMC-7721、L1210和HL60等3种癌细胞的抗增殖作用。结果:目标物的抗肿瘤活性高于母体化合物,尤其是苯环含有氟原子和硝基的化合物,其抗肿瘤作用与对照阿霉素相当。结论:噁二唑杂环可用于氟喹诺酮C-3羧基的等排体,被功能基侧链修饰有利于提高抗肿瘤活性。
基于抗菌氟喹诺酮羧酸(FQAs)药物的作用靶标拓扑异构酶也是抗肿瘤药物的重要作用靶点,且两种拓扑异构酶在功能和序列上具有相似性,因此可通过结构修饰将其抗菌活性转化为抗肿瘤活性,有望发展一类新结构的抗肿瘤药[1-2]。与此同时,有研究[3]已发现抗菌氟喹诺酮的C-3羧基并非是抗肿瘤活性所必需的药效团,可被其生物电子等排体如酰腙替代,这为FQAs由抗菌活性向抗肿瘤活性的转化提供了结构修饰新途径。然而,在众多的羧基等排体中,哪些是C-3羧基适宜的等排体,目前所知甚少。考虑到C-3羧基用唑稠杂环替代可产生抗肿瘤较好的C-3稠杂环化合物,表明C-3羧基可用(稠)杂环替代[4]。1,3,4-噁二唑杂环衍生物具有广泛的药理活性,在构建新药物分子中常作为重要的药效团骨架而受关注[5-6]。因此,该研究中试图将C-3羧基用1,3,4-噁二唑替代,并用功能酰腙基作为其修饰基,硫醚作为连接链,通过药效团的拼合,合成氟喹诺酮C-3噁二唑硫乙酰腙目标化合物,并对其初步的构效关系进行分析,为进一步结构优化提供指导。
1 材料与方法
1.1 合成路线 目标化合物7a~7j的制备路线图见图1。环丙沙星(化合物1)经肼解得环丙沙星酰肼(化合物2),与二硫化碳缩合得C-3酰肼二硫代甲酸(化合物3),在碱性水溶液中发生分子内环合反应得到C-3 1,3,4-噁二唑硫醇(化合物4),接着与氯乙酸乙酯进行亲核取代反应得C-3 1,3,4-噁二唑硫醚酯(化合物5),酯通过肼解得C-3 1,3,4-噁二唑硫乙酰肼(化合物6)。噁二唑硫乙酰肼与苯甲醛或取代的苯甲醛通过缩合反应得C-3噁二唑硫乙酰腙(化合物7a~7j )。
1.2 主要材料与仪器 环丙沙星为商业品,环丙沙星酰肼按文献[3]方法制备,其余试剂为分析纯。实验用金黄色葡萄球菌(S.aureusATCC-25923)和大肠埃希菌(E.coliATCC-25922)由河南大学淮河临床医学院检验科提供,人肝癌细胞(SMMC-7721)、小鼠白血病细胞(L1210)和人白血病细胞(HL60)购自中国科学院细胞库。WK-1B数字熔点仪(上海精密科学仪器厂); Bruker AM-400型核磁共振仪(瑞士Bruker公司),DMSO-d6为溶剂,TMS为内标; Esquire LC型质谱仪(德国Bruker公司); PE2400-Ⅱ型元素分析仪(美国PE 公司); BIO-AD-680型酶标仪(美国Bio-AD公司)。
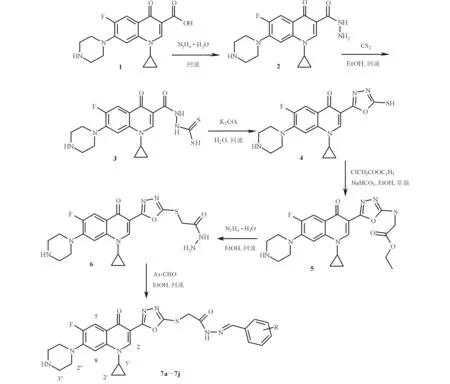
1~6:分别为化合物1~6;R:H(7a)、2-OH(7b)、2-CH3O(7c)、4-CH3O(7d)、3,4-(OCH2O)(7e)、3-CH3O-4-OH(7f)、3,4,5-(CH3O)3(7g)、4-F(7h)、4-Cl(7i)、4-O2N(7j)。图1 目标化合物7a~7j的制备路线图
1.3 化学合成 化合物1经肼解得化合物2后,将化合物2(20 g, 61.0 mmol)和二硫化碳(7.0 g,92.0 mmol)溶于300 mL无水乙醇中,加热回流反应12 h;热过滤产生的固体,醇洗涤,干燥,得淡黄色固体(化合物3)。粗品化合物3(10 g)悬浮于质量分数6%的碳酸钾水溶液(150 mL)中,加热回流反应10 h;加活性炭(1.0 g),回流脱色1 h。过滤,滤液用浓盐酸调pH为7.0, 滤集固体,去离子水洗涤,无水乙醇-DMF重结晶,得黄色固体(化合物4)。
将化合物4(10 g, 27.0 mmol)与碳酸氢钠(3.4 g, 41.0 mmol)在300 mL无水乙醇中回流反应溶解,滴加氯乙酸乙酯(4.0 g, 32.0 mmol),加热回流反应10 h;滤出固体,减压蒸除溶剂,残余物用无水乙醇重结晶,得无色固体(化合物5)。
将化合物5(10 g, 21.0 mmol)与质量分数80%水合肼(2.0 g, 32.0 mmol)在100 mL无水乙醇中回流反应12 h;冷却室温,滤集固体,用无水乙醇重结晶,得无色固体(化合物6)。取1.0 g(2.2 mmol)化合物6溶于20 mL无水乙醇中,加入等物质的量的苯甲醛或取代苯甲醛,回流反应6 h;室温放置,滤集析出的固体,冷乙醇洗涤。用无水乙醇或乙醇-DMF混合溶剂重结晶,得黄色固体目标化合物7a~7j。
1.4 体外抗菌活性实验 目标化合物7a~7j及母体环丙沙星配成128 mg/L的DMSO溶液作为供试样品,采用标准试管二倍稀释法测定其对金黄色葡萄球菌和大肠埃希菌的体外最低抑菌浓度(MIC)。
1.5 体外抗肿瘤活性实验 目标化合物7a~7j、对照蒽醌类抗肿瘤药阿霉素及母体环丙沙星用DMSO配成1.0×10-2mol/L的储备液,用RPMI 1640培养基稀释到所需浓度(0.1、1.0、10.0、50.0、100.0 mol/L)的供试液。①取对数生长期的SMMC-7721细胞,以每孔5 000个细胞接种于96孔板。隔夜培养后,加入供试液。48 h后弃去培养基, 每孔加入1 g/L MTT溶液100 μL, 继续培养4 h 后弃上清,每孔加入150 μL DMSO,轻轻振荡30 min,用酶标仪在570 nm波长处测OD值。②取对数生长期的L1210细胞和HL60细胞, 以每孔7 000个细胞接种于96孔板, 随后加入供试液。48 h后每孔加入5 g/L MTT溶液10 μL, 继续培养4 h 后加入10 g/L SDS溶液100 μL培养过夜, 用酶标仪在570 nm波长处测OD值。细胞生长抑制率=(1-实验组OD值/对照组OD值)×100%。以药物浓度对数值对细胞生长抑制率做线性回归得剂量-效应方程,依此计算出供试化合物对癌细胞的半数抑制浓度(IC50)。所有实验在相同条件下进行3次。
2 结果
2.1 化合物结构表征 1-环丙基-6-氟-7-哌嗪-1-基-3-(5-巯基-1,3,4-噁二唑-2-基)-喹啉(1H)-4-酮(化合物4):收率62%;熔点245~247 ℃;1H NMR(DMSO-d6)δ13.68(s,1H,SH),8.64(s,1H,2-H),7.86(d,J=13.2 Hz, 1H, 5-H),7.54(d,J=7.2 Hz, 1H, 8-H),3.68~3.76(m, 1H, 1′-H),3.36(t,J=6.2 Hz, 4H, 2×2″-H),2.66(t,J=6.2 Hz, 4H, 2×3″-H),1.20~1.32(m, 4H, 2×2′-H);EI-MS(m/z)计算值387.44,测定值388[M+H]+;元素分析(C18H18FN5O2S)计算值C 55.80%, H 4.68%, N 18.08%;测定值C 55.67%, H 4.52%, N 18.32%。
1-环丙基-6-氟-7-哌嗪-1-基-3-(5-乙氧甲酰乙硫基-1,3,4-噁二唑-2-基)-喹啉(1H)-4-酮(化合物5):收率76%;熔点165~167 ℃;1H NMR(DMSO-d6)δ8.66(s, 1H, 2-H),7.87(d,J=13.2 Hz, 1H, 5-H),7.63(d,J=7.2 Hz, 1H, 8-H),4.32,4.20(2s, 4H, OCH2和SCH2),3.68~3.74(m, 1H, 1′-H),3.35(t,J=6.2 Hz, 4H, 2×2″-H),2.64(t,J=6.2 Hz, 4H, 2×3″-H),1.21~1.27(m, 7H, CH3和2×2′-H);EI-MS(m/z)计算值473.53,测定值474[M+H]+;元素分析(C22H24FN5O4S)计算值C 55.80%, H 5.11%, N 14.79%;测定值C 55.94%, H 4.93%, N 15.04%。
1-环丙基-6-氟-7-哌嗪-1-基-3-(5-肼甲酰乙硫基-1,3,4-噁二唑-2-基)-喹啉(1H)-4-酮(化合物6):收率62%;熔点178~180 ℃;1H NMR(DMSO-d6)δ11.35(s, 1H, COH),8.68(s, 1H, 2-H),7.86(d,J=13.2 Hz, 1H, 5-H),7.64(d,J=7.2 Hz, 1H, 8-H),4.68(s, 2H, NH2),4.18(s, 2H, SCH2),3.66~3.75(m, 1H, 1′-H),3.36(t,J=6.2 Hz, 4H, 2×2″-H),2.68(t,J=6.2 Hz, 4H, 2×3″-H),1.22~1.25(m, 4H, 2×2′-H);EI-MS(m/z)计算值459.51,测定值460[M+H]+;元素分析(C20H22FN7O3S)计算值C 52.28%, H 4.83%, N 21.34%;测定值C 52.52%, H 4.68%, N 21.57%。
1-环丙基-6-氟-7-哌嗪-1-基-3-(5-苯甲叉肼甲酰乙硫基-1,3,4-噁二唑-2-基)-喹啉(1H)-4-酮(化合物7a):收率76%;熔点213~215 ℃;1H NMR(DMSO-d6)δ11.37(s, 1H, COH),8.73(s, 1H, 2-H),8.52(s, 1H, CH=N),7.85(d,J=13.2 Hz, 1H, 5-H),6.84~7.68(m, 6H, 8-H和Ph-H),4.20(s, 2H, SCH2),3.72~3.78(m, 1H, 1′-H),3.36(t,J=6.2 Hz, 4H, 2×2″-H),2.66(t,J=6.2 Hz, 4H, 2×3″-H),1.21~1.26(m, 4H, 2×2′-H);EI-MS(m/z)计算值547.62,测定值548[M+H]+;元素分析(C27H26FN7O3S)计算值C 59.22%, H 4.79%, N 17.90%;测定值C 59.46%, H 4.60%, N 18.14%。
1-环丙基-6-氟-7-哌嗪-1-基-3-[5-(2-羟基苯甲叉肼基)甲酰乙硫基-1,3,4-噁二唑-2-基]-喹啉(1H)-4-酮(化合物7b):收率64%;熔点 192~194 ℃;1H NMR(DMSO-d6)δ11.36(s, 1H, COH),9.68(s, 1H, OH),8.75(s, 1H, 2-H),8.62(s, 1H, CH=N),7.87(d,J=13.2 Hz, 1H, 5-H),7.15~7.72(m, 5H, 8-H和Ph-H),4.22(s, 2H, SCH2),3.70~3.76(m, 1H, 1′-H),3.37(t,J=6.2 Hz, 4H, 2×2″-H),2.74(t,J=6.2 Hz, 4H, 2×3″-H),1.23~1.27(m, 4H, 2×2′-H);EI-MS(m/z)计算值563.62,测定值564[M+H]+;元素分析(C27H26FN7O4S)计算值C 57.54%, H 4.65%, N 17.40%;测定值C 57.73%, H 4.50%, N 17.66%。
1-环丙基-6-氟-7-哌嗪-1-基-3-[5-(2-甲氧基苯甲叉肼基)甲酰乙硫基-1,3,4-噁二唑-2-基]-喹啉(1H)-4-酮(化合物7c):收率57%;熔点185~187 ℃;1H NMR(DMSO-d6)δ11.38(s, 1H, COH),8.76(s, 1H, 2-H),8.57(s, 1H, CH=N),7.88(d,J=13.2 Hz, 1H, 5-H),7.26~7.77(m, 5H, 8-H和Ph-H),4.23(s, 2H, SCH2),3.86(s, 3H, OCH3),3.74~3.78(m, 1H, 1′-H),3.38(t,J=6.2 Hz, 4H, 2×2″-H),2.76(t,J=6.2 Hz, 4H, 2×3″-H),1.26~1.32(m, 4H, 2×2′-H);EI-MS(m/z)计算值577.64,测定值578[M+H]+;元素分析(C28H28FN7O4S)计算值C 58.22%, H 4.89%, N 16.97%;测定值C 58.46%, H 4.73%, N 17.18%。
1-环丙基-6-氟-7-哌嗪-1-基-3-[5-(4-甲氧基苯甲叉肼基)甲酰乙硫基-1,3,4-噁二唑-2-基]-喹啉(1H)-4-酮(化合物7d):收率83%;熔点218~220 ℃;1H NMR(DMSO-d6)δ11.36(s, 1H, COH),8.78(s, 1H, 2-H),8.58(s, 1H, CH=N),7.89(d,J=13.2 Hz, 1H, 5-H),7.25~7.76(m, 5H, 8-H和Ph-H),4.24(s, 2H, SCH2),3.88(s, 3H, OCH3),3.70~3.76(m, 1H, 1′-H),3.36(t,J=6.2 Hz, 4H, 2×2″-H),2.75(t,J=6.2 Hz, 4H, 2×3″-H),1.28~1.36(m, 4H, 2×2′-H);EI-MS(m/z)计算值577.64,测定值578[M+H]+;元素分析(C28H28FN7O4S)计算值C 58.22%, H 4.89%, N 16.97%;测定值C 58.42%, H 4.76%, N 17.12%。
1-环丙基-6-氟-7-哌嗪-1-基-3-[5-(3,4-二氧亚甲基苯甲叉肼基)甲酰乙硫基-1,3,4-噁二唑-2-基]-喹啉(1H)-4-酮(化合物7e):收率91%;熔点234~236 ℃;1H NMR(DMSO-d6)δ11.38(s, 1H, COH),8.82(s, 1H, 2-H),8.64(s, 1H, CH=N),8.06(d,J=13.2 Hz, 1H, 5-H),7.56~7.87(m, 4H, 8-H和Ph-H),6.23(s, 2H, OCH2O),4.26(s, 2H, SCH2),3.74~3.77(m, 1H, 1′-H),3.38(t,J=6.2 Hz, 4H, 2×2″-H),2.78(t,J=6.2 Hz, 4H, 2×3″-H),1.23~1.35(m, 4H, 2×2′-H);EI-MS(m/z)计算值591.63,测定值592[M+H]+;元素分析(C28H26FN7O5S)计算值C 56.85%, H 4.43%, N 16.57%;测定值C 56.68%, H 4.28%, N 16.83%。
1-环丙基-6-氟-7-哌嗪-1-基-3-[5-(3-甲氧-4-羟基-苯甲叉肼基)甲酰乙硫基-1,3,4-噁二唑-2-基]-喹啉(1H)-4-酮(化合物7f):收率93%;熔点245~247 ℃;1H NMR(DMSO-d6)δ11.37(s, 1H, COH),10.36(s, 1H, OH),8.78(s, 1H, 2-H),8.62(s, 1H, CH=N),7.94(d,J=13.2 Hz, 1H, 5-H),7.46~7.82(m, 4H, 8-H和Ph-H),4.25(s, 2H, SCH2),3.86(s, 3H, CH3O),3.70~3.78(m, 1H, 1′-H),3.36(t,J=6.2 Hz, 4H, 2×2″-H),2.72(t,J=6.2 Hz, 4H, 2×3″-H),1.25~1.33(m, 4H, 2×2′-H);EI-MS(m/z)计算值593.64,测定值594[M+H]+;元素分析(C28H28FN7O5S)计算值C 56.65%, H 4.75%, N 16.52%;测定值C 56.87%, H 4.52%, N 16.77%。
1-环丙基-6-氟-7-哌嗪-1-基-3-[5-(3,4,5-三甲氧苯甲叉肼基)甲酰乙硫基-1,3,4-噁二唑-2-基]-喹啉(1H)-4-酮(化合物7g):收率77%;熔点196~198 ℃;1H NMR(DMSO-d6)δ11.36(s, 1H, COH),8.76(s, 1H, 2-H),8.57(s, 1H, CH=N),7.88(d,J=13.2 Hz, 1H, 5-H),7.64(d,J=7.2 Hz,1H, 8-H),7.56(s, 2H, Ph-H),4.22(s, 2H, SCH2),3.88, 3.85(2s, 9H, 3×CH3O), 3.68~3.76(m, 1H, 1′-H),3.37(t,J=6.2 Hz, 4H, 2×2″-H),2.68(t,J=6.2 Hz, 4H, 2×3″-H),1.20~1.32(m, 4H, 2×2′-H);EI-MS(m/z)计算值637.70,测定值638[M+H]+;元素分析(C30H32FN7O6S)计算值C 56.51%, H 5.06%, N 15.38%;测定值C 56.74%, H 4.87%, N 15.63%。
1-环丙基-6-氟-7-哌嗪-1-基-3-[5-(4-氟苯甲叉肼基)甲酰乙硫基-1,3,4-噁二唑-2-基]-喹啉(1H)-4-酮(化合物7h):收率82%;熔点215~217 ℃;1H NMR(DMSO-d6)δ11.38(s, 1H, COH),8.84(s, 1H, 2-H),8.62(s, 1H, CH=N),8.16(d,J=7.5 Hz, 2H, Ph-H),8.06(d,J=13.2 Hz, 1H, 5-H),7.74~7.83(m, 3H, 8-H和Ph-H),4.26(s, 2H, SCH2),3.75~3.81(m, 1H, 1′-H),3.38(t,J=6.2 Hz, 4H, 2×2″-H),2.74(t,J=6.2 Hz, 4H, 2×3″-H),1.26~1.35(m, 4H, 2×2′-H);EI-MS(m/z)计算值565.61,测定值566[M+H]+;元素分析(C27H25F2N7O3S)计算值C 57.34%, H 4.46%, N 17.33%;测定值C 57.56%, H 4.22%, N 17.54%。
1-环丙基-6-氟-7-哌嗪-1-基-3-[5-(4-氯苯甲叉肼基)甲酰乙硫基-1,3,4-噁二唑-2-基]-喹啉(1H)-4-酮(化合物7i):收率67%;熔点183~185 ℃;1H NMR(DMSO-d6)δ11.36(s, 1H, COH),8.80(s, 1H, 2-H),8.57(s, 1H, CH=N),8.01(d,J=13.2 Hz, 1H, 5-H),7.68~7.85(m, 3H, 8-H和Ph-H),4.23(s, 2H, SCH2),3.73~3.78(m, 1H, 1′-H),3.36(t,J=6.2 Hz, 4H, 2×2″-H),2.70(t,J=6.2 Hz, 4H, 2×3″-H),1.21~1.33(m, 4H, 2×2′-H);EI-MS(m/z)计算值582.02,测定值582[M+H]+;元素分析(C27H25ClFN7O3S)计算值C 55.72%, H 4.33%, N 16.84%;测定值C 55.95%, H 4.13%, N 16.72%。
1-环丙基-6-氟-7-哌嗪-1-基-3-[5-(4-硝基苯甲叉肼基)甲酰乙硫基-1,3,4-噁二唑-2-基]-喹啉(1H)-4-酮(化合物7j):收率74%;熔点242~246 ℃;1H NMR(DMSO-d6)δ11.45(s, 1H, COH),8.87(s, 1H, 2-H),8.66(s, 1H, CH=N),8.15(d,J=13.2 Hz, 1H, 5-H),8.08(d,J=7.5 Hz, 2H, Ph-H),7.73~7.88(m, 3H, 8-H和Ph-H),4.26(s, 2H, SCH2),3.76~3.81(m, 1H, 1′-H),3.38(t,J=6.2 Hz, 4H, 2×2″-H),2.74(t,J=6.2 Hz, 4H, 2×3″-H),1.25~1.36(m, 4H, 2×2′-H);EI-MS(m/z)计算值592.61,测定值593[M+H]+;元素分析(C27H25FN8O5S)计算值C 54.72%, H 4.25%, N 18.91%;测定值C 54.92%, H 4.03%, N 19.15%。
2.2 体外抗菌活性实验结果 目标化合物7a~7j对两种实验细菌株的MIC为64.00 mg/L,而对照环丙沙星的MIC为0.25 mg/L。
2.3 体外抗肿瘤活性实验结果 见表1。
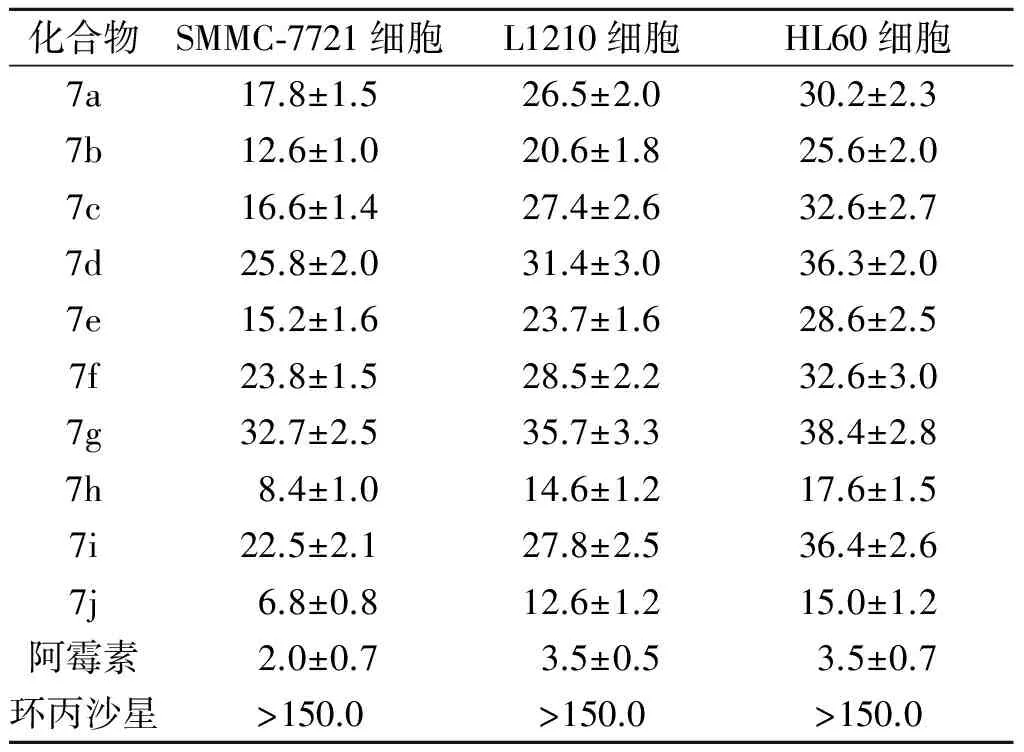
表1 化合物7a~7j对SMMC-7721、L1210和HL60细胞的IC50(n=3) mol/L
3 讨论
环丙沙星不经酯化可与水合肼直接发生肼解反应得到环丙沙星酰肼。酰肼与二硫化碳不需要碱催化可发生缩合得到环丙沙星酰肼二硫代甲酸,接着在碳酸钾碱性水溶液中发生分子内的加成和消除一分子的H2S形成C-3噁二唑硫醇,它与氯乙酸酯发生亲核取代反应得到C-3 噁二唑硫醚酯,再通过酯的肼解得到C-3噁二唑硫醚酰肼,进一步与苯甲醛或取代的苯甲醛缩合可得目标化合物。
体外抗菌活性实验结果表明,10个目标化合物7a~7j对两种实验细菌株的MIC为64.00 mg/L,而对照环丙沙星的MIC为0.25 mg/L,这进一步证实C-3羧基是抗菌活性所必需的药效团。与此同时,体外抗肿瘤活性实验结果表明,10个目标化合物对3种肿瘤细胞株的IC50均低于原药环丙沙星,表明噁二唑杂环作为C-3羧基的等排体可显著提高其抗肿瘤活性。初步的构效关系表明,当苯环带有吸电子基的氟原子或硝基时,其化合物的抗肿瘤活性高于其他取代基化合物的活性。提示含吸电子基的苯环可活化与其相连的席夫碱亚胺双键,有利于与靶点发生亲核反应而产生细胞毒活性。基于此,氟喹诺酮C-3羧基并非是抗肿瘤活性所必要的药效团,可被唑杂环生物电子等排体替代,且用功能基侧链修饰优化有利于提高其抗肿瘤活性,这为进一步的结构优化提供了新的思路和方法。
[1]ALDRED KJ,SCHWANZ HA,LI G,et al.Activity of quinolone CP-115,955 against bacterial and human type Ⅱ topoisomerases is mediated by different interactions[J].Biochemistry,2015,54(5):1278
[2]POMMIER Y.Drugging topoisomerases: lessons and challenges[J].ACS Chem Biol,2013,8(1):82
[3]尚慧杰,高留州,谢玉锁,等.N-甲基环丙沙星酰腙的合成及抗肿瘤活性[J].郑州大学学报(医学版),2015,50(5):597
[4]YOU QD,LI ZY,HUANG CH,et al.Discovery of a novel series of quinolone and naphthyridine derivatives as potential topoisomerase Ⅰ inhibitors by scaffold modification[J].J Med Chem,2009,52(18):5649
[5]GAMAL EL-DIN MM,EL-GAMAL MI,ABDEL-MAKSOUD MS,et al.Synthesis and in vitro antiproliferative activity of new 1,3,4-oxadiazole derivatives possessing sulfonamide moiety[J].Eur J Med Chem,2015,90:45
[6]VALENTE S,TRISCIUOGLIO D,DE LUCA T,et al.1,3,4-oxadiazole-containing histone deacetylase inhibitors: anticancer activities in cancer cells[J].J Med Chem,2014,57(14):6259
(2016-04-15收稿 责任编辑姜春霞)
Synthesis and antitumor activity detection of fluoroquinolone C-3 oxadiazole sulfanylacetylhydrazones
LIKe1),ZHANGHuili1),WENJing2),HUGuoqiang2),HUANGWenlong3)
1)SchoolofPharmacy,ZhengzhouUniversityofIndustrialTechnology,Zhengzhou451150 2)SchoolofPharmacy,HenanUniversity,Kaifeng,Henan475001 3)CenterofDrugDiscovery,ChinaPharmaceuticalUniversity,Nanjing210009
fluoroquinolone; oxadiazole; acylhydrazone; bioisostere; antitumor activity
Aim: To discover an efficient strategy for a conversion of antibacterial fluoroquinolones into antitumor fluoroquinolones. Methods: Ten novel fluoroquinolone C-3 oxadiazole sulfanylacetylhydrazone derivatives were designed and synthesized with an oxadiazole ring as the C-3 bioisostere modified by a functionalized sulfanylacetylhydrazone side-chain from ciprofloxacin, respectively. The structures of the title compounds were characterized by elemental analysis and spectral data, and theinvitroantitumor activity against SMMC-7721, L1210 and HL60 cell lines was evaluated by MTT assay. Results: The title compounds demonstrated more antiproliferative activity than the parent. In particular, compounds bearing a fluorine atom or nitro group attached to benzene ring were comparable to the control doxorubicin. Conclusion: An azole modified with functionalized side-chain as the bioisosteric replacement of the C-3 carboxylic group is favorable to improvement of antitumor activity.
10.13705/j.issn.1671-6825.2017.01.008
*国家自然科学基金面上资助项目 20872028,21072045
R979.1
猜你喜欢
化工技术与开发(2022年11期)2022-11-29
湘潭大学自然科学学报(2022年2期)2022-07-28
林业工程学报(2022年1期)2022-02-26
中国饲料(2021年17期)2021-11-02
北京大学学报(自然科学版)(2021年3期)2021-07-16
烟台大学学报(自然科学与工程版)(2021年1期)2021-03-19
浙江中西医结合杂志(2020年9期)2020-09-22
看世界·学术下半月(2020年5期)2020-09-10
当代化工(2020年8期)2020-09-09
分析化学(2015年7期)2015-07-30
