
高效液相色谱-三重四极杆质谱法同时测定新型“香料”毒品中的10种合成大麻素
2016-12-20 02:53张春水翟晚枫
分析测试学报 2016年3期
张春水,翟晚枫
(公安部物证鉴定中心,北京 100038)
高效液相色谱-三重四极杆质谱法同时测定新型“香料”毒品中的10种合成大麻素
张春水,翟晚枫*
(公安部物证鉴定中心,北京 100038)
为测定新型“香料”毒品中常见的合成大麻素成分,研究开发了高效液相色谱-三重四极杆质谱联用分析方法。采用安捷伦Poroshell 120 EC-C18(3.0 mm×50 mm,2.7 μm) 色谱柱,以高纯水-甲醇作流动相进行梯度洗脱,柱温30 ℃,流速0.3 mL/min。采用电喷雾电离-正离子(ESI+)、负离子(ESI-)分段检测模式,并对合成大麻素的质谱特征和离子碎裂规律进行研究。结果表明,采用该方法可以实现对常见10种合成大麻素的定性和定量分析,正、负离子模式下检测的目标物分别在1~100,10~1 000 ng/mL范围内呈良好线性,日内相对标准偏差(RSD)均不大于3.2%,日间RSD均不大于6.3%。经加标回收率测定和实际样本检验,该方法快速、准确、灵敏、可靠,适用于新型“香料”毒品中常见合成大麻素成分的定性定量检测。
高效液相色谱-质谱联用法;香料;毒品;合成大麻素
合成大麻素(Synthetic cannabinoids)和拟大麻素(Cannabimimetics)常被通称为合成大麻素,前者是与大麻活性成分四氢大麻酚(Tetrahydrocannabinol,Δ9-THC)结构相似的化合物,后者在化学结构上与四氢大麻酚并不相似,但其在体内的生理药理作用与之类似。这两类物质均为与内源性大麻素系统相作用的大麻素受体激动剂,很多情况下,它们比大麻植物的主要精神活性成分Δ9-THC具有更强的生理和药理作用[1]。合成大麻素大多数以治疗为目的由科学家合成得到,后被地下制毒工厂迅速发展为新型毒品,作为大麻的替代品而滥用[2]。制毒者通常将合成大麻素溶液喷涂于植物基质上,晾干后分装出售,这种新型毒品常被称作“香料(Spice)”。
广义的合成大麻素主要包含萘吲哚类、萘甲基吲哚类、萘吡咯类、萘甲基茚类、苯乙基吲哚类、环己基苯酚类和经典大麻素类7类结构[3]。1994年,美国化学家Huffman等[4]研制了以萘甲酰基吲哚类为主的系列化合物,即最早的合成大麻素,后被命名为JWH系列化合物,包括JWH-018、JWH-073和JWH-200等。此后,HU-210、CP47,497、CP47,497(C8)等不同种类的合成大麻素陆续被研制成功并迅速流行于国外。2010年我国大陆警方查获、检验并公开报道了第一例“PeaceOut”[5]和“Spike 99”香料毒品案例[6]。
对于合成大麻素类物质,主要的检验方法有气相色谱-质谱法[7-9]、液相色谱法[10]、液相色谱-质谱法[11-14]、核磁共振波谱法[15]等,而我国目前仅有气相色谱-质谱法[5-6,16-18]和高效液相色谱法[19-22]的文献报道。2014年,本课题组建立了高效液相色谱法同时定量测定新型“香料”毒品中10种合成大麻素的检验方法[22],其后,考虑到LC-MS/MS方法具有灵敏度高、准确性好、能够弥补液相色谱法定性功能不足以及缩短分析时间等特点,也为进一步填补我国采用LC-MS/MS方法检验合成大麻素类物质的研究空白,本文又开发了检测常见10种合成大麻素的高效液相色谱-三重四极杆质谱(HPLC-MS/MS)定性、定量分析方法,并依据质谱特征推测了其碎裂途径。
1 实验部分
1.1 仪器与试剂
Agilent 1290 高效液相色谱仪;Agilent 6460 QQQ三重四极杆质谱仪;瑞士Mettler十万分之一电子天平;美国Millipore纯水仪。
目标分析物为常见的10种合成大麻素。其中JWH-018(1-戊基-3-(1-萘甲酰基)吲哚,1.0 mg/mL)、JWH-073(1-丁基-3-(1-萘甲酰基)吲哚,1.0 mg/mL)、JWH-200([1-[2-(4-吗啉基)乙基]-1H-吲哚-3-基]-1-萘基甲酮,1.0 mg/mL)、CP47,497(5-(1,1-二甲基庚基)-2-[(1R,3S)-3-羟基环己基]苯酚,1.0 mg/mL)、CP47,497(C8) (5-(1,1-二甲基辛基)-2-[(1R,3S)-3-羟基环己基]苯酚,1.0 mg/mL)、HU-210((1,1-二甲基庚基)-6a,7,10,10a-四氢-1-羟基-6,6-二甲基-6H-二苯并[b,d]吡喃-9-甲醇,1.0 mg/mL)标准品溶液购于美国Cerilliant公司;JWH-147((1-己基-5-苯基-1H-吡咯-3-基)(萘-1-基)甲酮,纯度大于97%)、JWH-203(1-戊基-3-(2-氯苯乙酰基)吲哚,纯度大于98%)标准品购于加拿大TRC公司;JWH-250(2-(2-甲氧基苯基)-1-(1-戊基-1H-吲哚-3-基)乙酮,纯度大于98%)、JWH-122(1-戊基-3-(4-甲基-1-萘甲酰基)吲哚,纯度大于98%)为实验室自合成对照品。甲醇(美国Thermo Fisher公司)。“香料”毒品样本来源于案件中收缴或人工模拟。
1.2 色谱-质谱条件
安捷伦Poroshell 120 EC-C18(3.0 mm×50 mm,2.7 μm) 色谱柱;流动相:高纯水(A)和甲醇(B);梯度洗脱:0~12 min,70%~95%B;柱温30 ℃;流速0.3 mL/min。
采用电喷雾电离-正离子模式(ESI+)和负离子模式(ESI-),雾化气温度350 ℃,雾化气压力30 psi,干燥气流量10 L/min,正离子模式下毛细管电压4 000 V,负离子模式下毛细管电压5 000 V,加速电压3 V。
1.3 标准储备液与混合标准溶液的配制
精密称取各标准品粉末或量取各标准品溶液适量,用甲醇定容于10 mL容量瓶,得到浓度为1.0 μg/mL的混合标准储备液。取各标准物质适量,用甲醇稀释定容,配制成浓度分别为1.0~1 000.0 ng/mL的系列标准溶液备用。
1.4 样品溶液的制备
定性分析时将收缴的“香料”毒品样本充分研磨,精密称取2.0 mg,加入50 mL甲醇,超声5 min,经0.22 μm滤膜过滤,取1.5 mL进样。
定量分析时需制备平行双样,每个样品需经过两次稀释,如精密称取各30.0 mg的两份样品,分别加入40 mL甲醇,超声5 min,经0.22 μm滤膜过滤后取1 mL,再加入40 mL甲醇,振摇均匀后取1.5 mL进样。
2 结果与讨论
2.1 色谱-质谱条件的优化
2.1.1 流动相的选择 比较了流动相分别为水-甲醇、0.2%甲酸-甲醇、5 mmol/L甲酸铵-甲醇和(0.2%甲酸-5 mmol/L甲酸铵)-甲醇时的离子响应,发现添加甲酸后,10种大麻素的离子响应均被抑制;添加甲酸铵后,多数目标物的离子响应出现不同程度的降低。对于采用正离子检测模式的物质,添加甲酸铵与同时添加酸和甲酸铵的离子响应差异不大。而对于采用负离子检测模式的物质(CP47,497、CP47,497(C8)、HU-210),同时添加酸和甲酸铵会降低离子响应。可见水-甲醇体系能够获得最佳的离子响应,但考虑到该体系无缓冲作用,又对4种体系的精密度进行了考察。结果表明,4种体系的日内和日间精密度无显著差异。因此,选择水-甲醇体系作为流动相。

图1 优化条件下10种合成大麻素的HPLC-MS/MS的MRM色谱图Fig.1 HPLC-MS/MS MRM chromatogram of 10 synthetic cannabinoids under optimized conditions
2.1.2 有机相初始浓度与梯度陡度的影响 单因素实验结果表明,在梯度洗脱程序中,当流动相有机相梯度陡度增加或初始浓度增加时,离子响应增加,保留时间缩短,分离度降低。另一方面,10种目标物中,有7种物质采用正离子模式检测,而另外3种采用负离子模式检测,需要一定的分离度。综合考虑以上因素,将梯度洗脱程序参数确定为:有机相(甲醇)初始浓度70%,梯度陡度1.6%/min,柱温30 ℃,流速0.3 mL/min。2.1.3 质谱参数的确定 10种合成大麻素的子离子、Fragmentor 电压和CE值优化结果见表1。离子源参数部分,采用单因素实验的方法对毛细管电压(Capillary voltage)、干燥气流量(Gas flow)、干燥气温度(Gas temperature)和雾化气压力(Nebulizer)进行优化。积分后,将峰面积做归一化处理并作图。结果表明,正离子模式下检测的7种物质的质谱响应先随毛细管电压的增大而增大,当毛细管电压升至4 000 V左右时质谱响应开始下降;而负离子模式下检测的3种物质的质谱响应则随毛细管电压的升高而不断升高(3 000~5 000 V范围内)。在干燥气流量为10 L/min左右时,各目标物的质谱响应达到最大值(6~12 L/min范围内)。此外,10种目标物的质谱响应随干燥气温度的增加而增加(200~350 ℃范围内),随雾化气压力的升高而降低(20~50 psi范围内)。优化条件下,10种合成大麻素的MRM色谱图如图1所示。
表1 10种合成大麻素的HPLC-MS/MS采集参数
Table 1 HPLC-MS/MS acquisition parameters for 10 synthetic cannabinoids
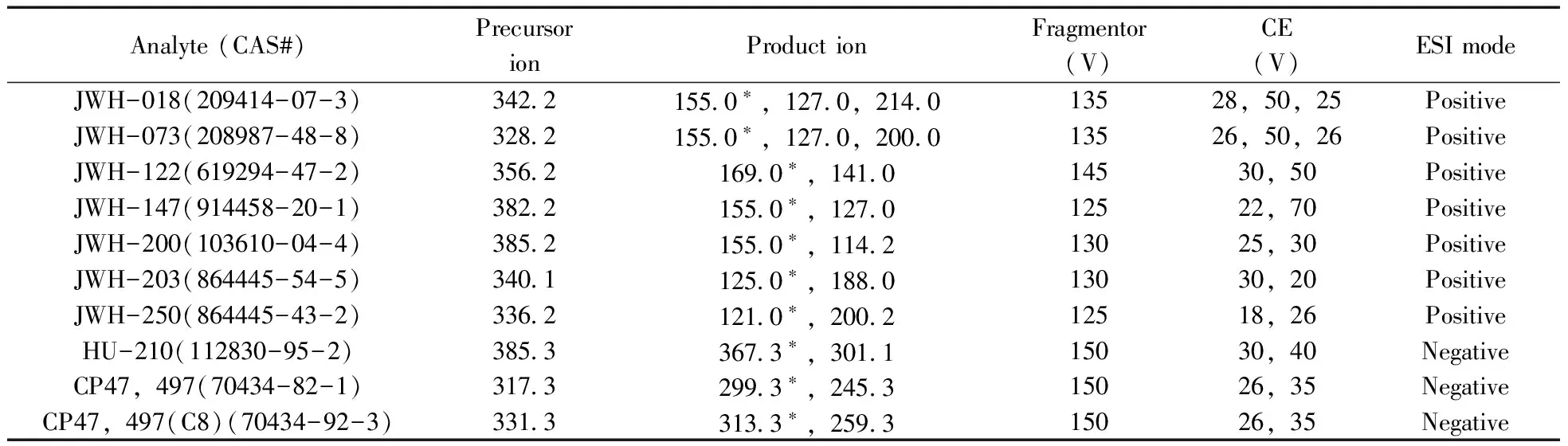
Analyte(CAS#)PrecursorionProductionFragmentor(V)CE(V)ESImodeJWH-018(209414-07-3)34221550∗,1270,214013528,50,25PositiveJWH-073(208987-48-8)32821550∗,1270,200013526,50,26PositiveJWH-122(619294-47-2)35621690∗,141014530,50PositiveJWH-147(914458-20-1)38221550∗,127012522,70PositiveJWH-200(103610-04-4)38521550∗,114213025,30PositiveJWH-203(864445-54-5)34011250∗,188013030,20PositiveJWH-250(864445-43-2)33621210∗,200212518,26PositiveHU-210(112830-95-2)38533673∗,301115030,40NegativeCP47,497(70434-82-1)31732993∗,245315026,35NegativeCP47,497(C8)(70434-92-3)33133133∗,259315026,35Negative
*quantitative ion
2.2 离子碎裂规律与质谱特征
10种合成大麻素中,JWH-018、JWH-073、JWH-200、JWH-122是萘甲酰基吲哚类的合成大麻素,JWH-147的结构也与之相似,含有萘甲酰基吡咯结构。通过质谱解析得出这一类萘甲酰基类合成大麻素离子碎裂的过程如下:首先化合物分子在溶剂和电场的作用下形成[M+1]+的准分子离子,进而带正电荷的羰基诱导羰基和吲哚环(或吡咯环)之间或羰基和萘环之间发生断裂产生离子碎片。其中JWH-018、JWH-073、JWH-147能够产生质量数为m/z155.0和127.0的碎片离子。在定性分析方面,除可通过子离子丰度比和保留时间增加这3种物质的区分度外,还可为JWH-018、JWH-073分别增加1对定性离子(m/z214.0和200.0),均由羰基和萘环连接的化学键断裂所产生,亦即分子离子断裂为除萘环外的另一部分的质量数,也是比较稳定且响应较强的碎片。
JWH-203是苯乙酰基吲哚类合成大麻素,二级碎片同样来自于羰基两侧的α键断裂;JWH-250结构和碎片略显特殊,主要产生m/z121.0和200.2两个碎片离子,其中m/z121.0可能是羰基碳与吲哚环断裂后发生羰基脱氧而形成,m/z200.2则可能是羰基碳与苯环发生断裂后再发生羰基脱氧而形成。
CP47,497和CP47,497(C8)属于环己基苯酚类,碎片来源于失去环羟基或六元环的断裂;HU-210属于经典大麻素类,碎片来源于失去羟基或季碳与长链间的化学键断裂。10种合成大麻素的质谱图和推测碎裂途径见图2。


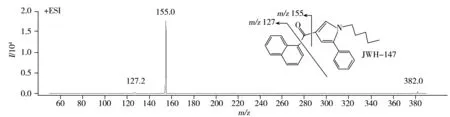

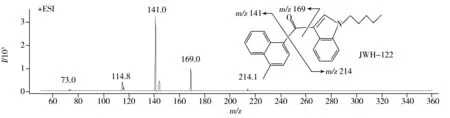
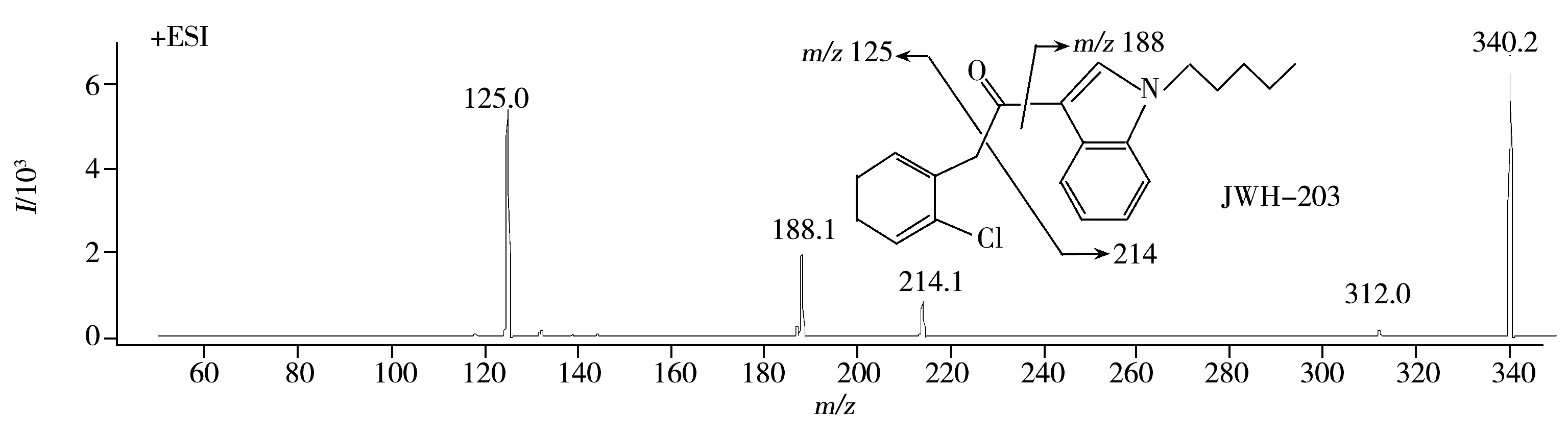


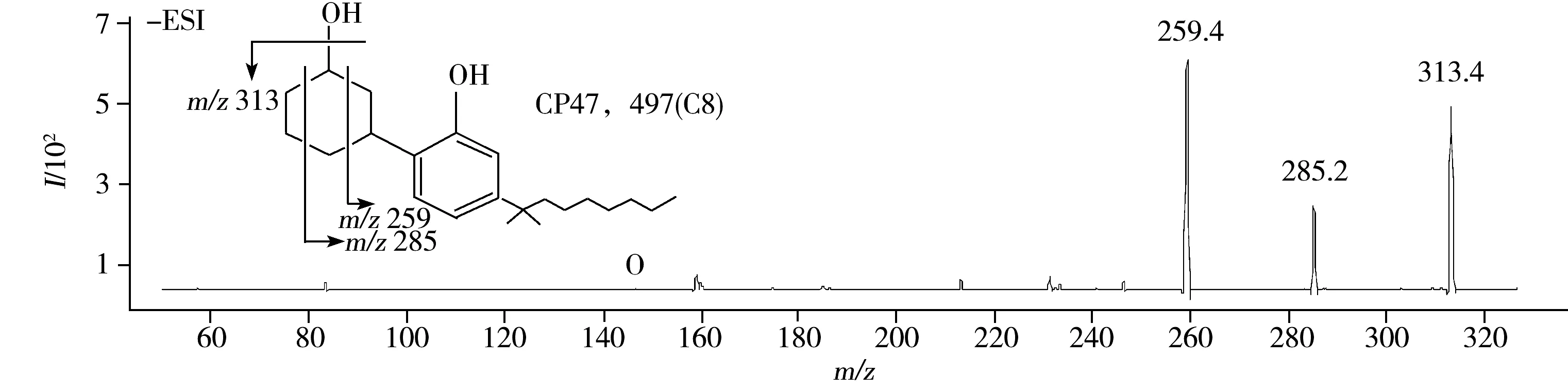
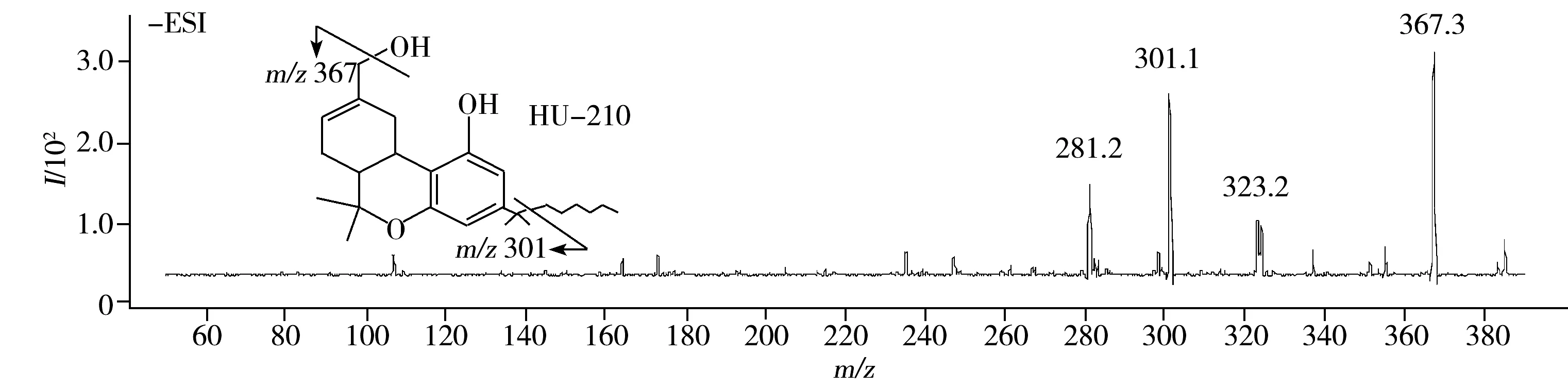
图2 10种合成大麻素的子离子质谱图和推测碎裂途径
Fig.2 HPLC-MS/MS product ion spectra and proposed fragmentations of 10 synthetic cannabinoids
2.3 方法评价
2.3.1 线性关系及检出限 将“1.3”方法制备的系列混合标准溶液,按优化的色谱-质谱条件进行分析并计算线性回归方程。正离子模式下检测的7种物质的线性范围为1~100 ng/mL,负离子模式下检测的3种物质的线性范围为10~1 000 ng/mL,相关系数(r)均大于0.999 0,结果见表2。结果表明,方法线性关系良好,具有较高灵敏度,检出限LOD(S/N=3)为0.010~0.200 ng·mL-1。
表2 10种合成大麻素的线性回归方程、相关系数及检出限
Table 2 Regression equations,correlation coefficients(r) and detection limits ( LODs) of 10 synthetic cannabinoids

CompoundLinearrangeρ/(ng·mL-1)RegressionequationrLODρ/(ng·mL-1)JWH-2001~100Y=303185X+103071099920010JWH-2501~100Y=753217X+112332099950025JWH-0731~100Y=528561X+156068099910010JWH-2031~100Y=263218X+126895099930015JWH-0181~100Y=599121X+107833099920020JWH-1221~100Y=773663X+152845099970010
(续表2)

CompoundLinearrangeρ/(ng·mL-1)RegressionequationrLODρ/(ng·mL-1)CP47,49710~1000Y=27222X-15288099950100JWH-1471~100Y=752181X+139111099930010CP47,497(C8)10~1000Y=30255X-20926099920100HU-21010~1000Y=16996X-11987099930200
2.3.2 加标回收率与精密度 以JWH-122和JWH-250作为实验对象,分别选择高、低两种含量的样品,精密称取每种已知含量的样品3份,分别加入约为已知含量50%,100%,150%的标准品,采用本方法进行分析,每个浓度重复测定3次,计算回收率。结果测得加标回收率为97.9%~103.1%,相对标准偏差(RSD)为1.2%~2.6%(表3),方法不存在系统性偏差。
表3 JWH-122和JWH-250的加标回收率与相对标准偏差
Table 3 Spiked recoveries and RSDs of JWH-122 and JWH-250
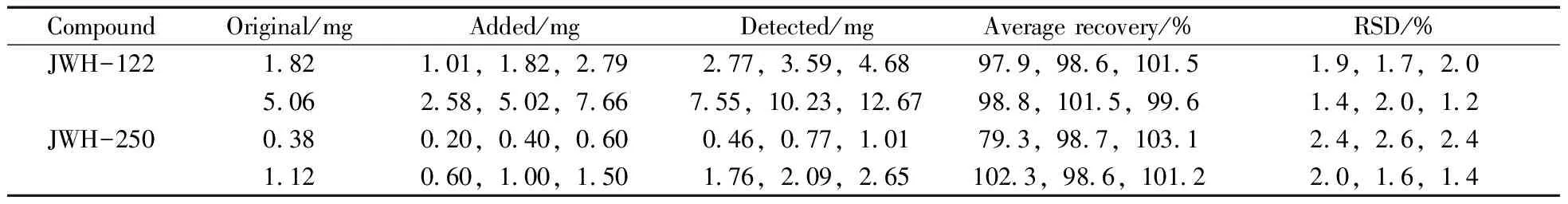
CompoundOriginal/mgAdded/mgDetected/mgAveragerecovery/%RSD/%JWH-122182101,182,279277,359,468979,986,101519,17,20506258,502,766755,1023,1267988,1015,99614,20,12JWH-250038020,040,060046,077,101793,987,103124,26,24112060,100,150176,209,2651023,986,101220,16,14
对于JWH-200等7种正离子模式下检测的目标物,制备浓度分别为100.0,10.0,2.0 ng/mL的混合标准品溶液各6份,对CP47,497等3种负离子模式下检测的目标物,制备浓度分别1 000.0,100.0,20.0 ng/mL的混合标准品溶液各6份。每日伴随标准曲线检测,计算含量,得到日内RSD;连续测量6 d,计算得到日间RSD(见表4)。结果显示,10种合成大麻素的日内RSD均不大于3.2%,日间RSD均不大于6.3%,表明方法的精密度良好。
表4 10种合成大麻素的日内、日间相对标准偏差
Table 4 Intra- and inter-RSDs of 10 synthetic cannabinoids
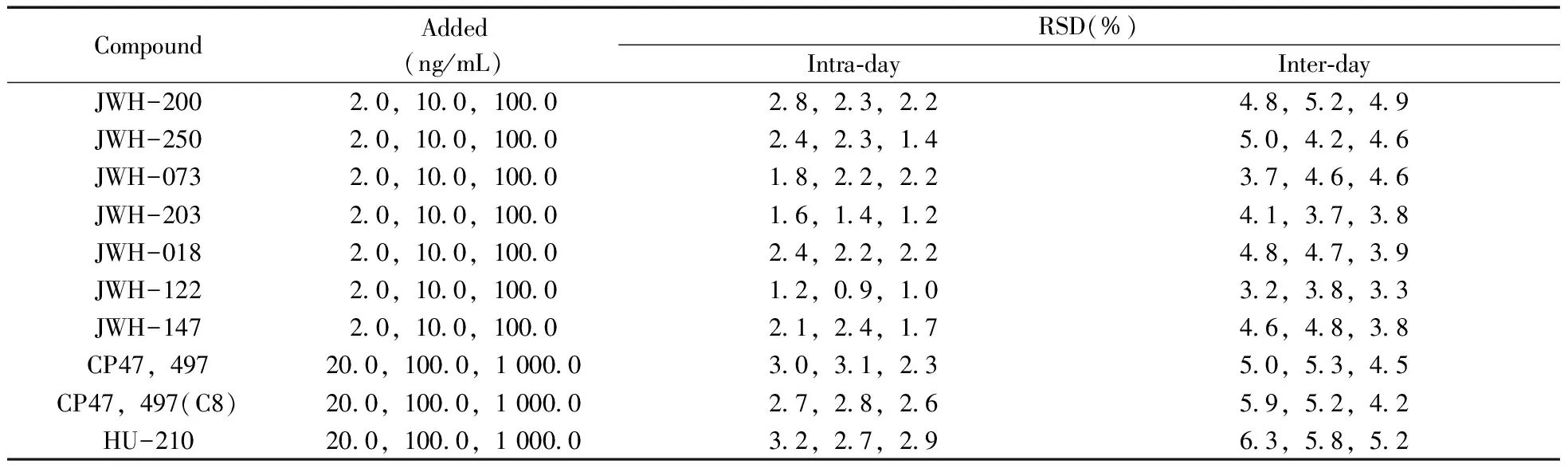
CompoundAdded(ng/mL)RSD(%)Intra⁃dayInter⁃dayJWH-20020,100,100028,23,2248,52,49JWH-25020,100,100024,23,1450,42,46JWH-07320,100,100018,22,2237,46,46JWH-20320,100,100016,14,1241,37,38JWH-01820,100,100024,22,2248,47,39JWH-12220,100,100012,09,1032,38,33JWH-14720,100,100021,24,1746,48,38CP47,497200,1000,1000030,31,2350,53,45CP47,497(C8)200,1000,1000027,28,2659,52,42HU-210200,1000,1000032,27,2963,58,52
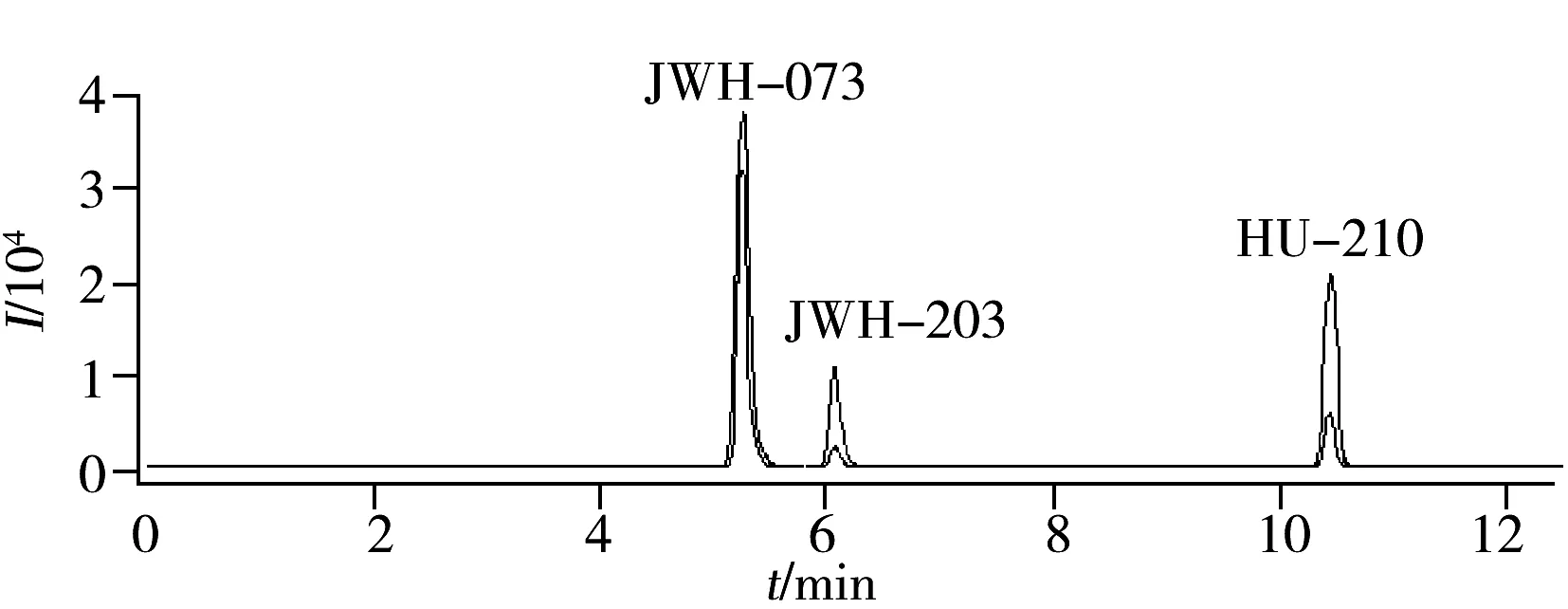
图3 某“香料”毒品样本的提取离子流色谱图Fig.3 HPLC-MS/MS MRM chromatogram of the “spice” sample
2.4 实际样品分析
采用“1.4”样品处理方法及“1.2”色谱-质谱条件对“香料”毒品样品进行分析,结果均显示色谱和质谱行为良好,方法稳定、可靠。以其中某一草叶状样品为例,经筛查,确定样品中含有JWH-073、JWH-203和HU-210 3种合成大麻素(样品与标准品的色谱峰保留时间和相对离子丰度比均一致)。定量计算,得到3种目标物含量分别为0.33%,0.17%,4.36%,各目标物的分离效果好、抗干扰性强,该样品的MRM色谱图如图3所示。
3 结 论
本文建立了高效液相色谱-质谱联用法同时测定新型“香料”毒品中常见10种合成大麻素的定性、定量方法,并优化了液相色谱-质谱参数。该法已经过方法学验证和实际案例样本的检验,显示出很好的应用前景。通过质谱解析对10种合成大麻素离子的裂解过程进行了研究,推测出可能的离子碎裂途径,可为未知“香料”毒品样本的检验和新毒品的发现提供借鉴。
[1] Vardakou I,Pistos C,Spiliopoulou C.Toxicol.Lett.,2010,197(3):157-162.
[2] ElSohly M A,Gul W,Wanas A S,Radwan M M.WanasLifeSciences,2014,97(1):78-90.
[3] Dowling G,Regan L.J.Chromatogr.B,2011,879(3/4):253-259.
[4] Huffman J W,Dai D,Martin B R,Compton D R.Bioorg.Med.Chem.Lett.,1994,4(4): 563-566.
[5] Xu P,Liu K L,Qian Z H.ChineseJournalofDrugAbusePreventionandTreatment(徐鹏,刘克林,钱振华.中国药物滥用防治杂志),2012,18(2):120-123.
[6] Xu P,Wang Y,Qian Z H,Zheng H,Liu K L.ChineseJournalofDrugDepend(徐鹏,王一,钱振华,郑珲,刘克林.中国药物依赖性杂志),2011,20(1): 47-49.
[7] Gregori A,Damiano F,Bonavia M,Mileo V,Varani F,Monfreda M.ScienceandJustice,2013,53(3):286-292.[8] Kavanagh P,Grigoryev A,Melnik A,Savchuk S,Simonov A,Rozhanets V.J.Chromatogr.B,2013,934:102-108.[9] Emerson B,Durham B,Gidden J,Lay Jr J D.J.ForensicSci.Int.,2013,229(1/3):1-6.
[10] Merola G,Aturki Z,D'Orazio G,Gottardo R,Macchia T,Tagliaro F,Fanali S.J.Pharm.Biomed.Anal.,2012,71:45-53.
[11] Jager A D,Warner J V,Henman M,Ferguson W,Hall A.J.Chromatogr.B,2012,897:22-31.
[12] Lovett D P,Yanes E G,Herbelin T W,Knoerzer T A,Levisky J A.ForensicSci.Int.,2013,226(1/3):81-87.
[13] Adamowicz P,Zuba D,Sekula K.ForensicSci.Int.,2013,233(1/3):320-327.
[14] Scheidweiler K B,Huestis M A.J.Chromatogr.A,2014,1327:105-117.
[15] Moosmann B,Kneisel S,Girreser U,Brecht V,Westphal F,Auwärter V.ForensicSci.Int.,2012,220(1/3):e17-e22.[16] Wu Z P,Zheng S Q,Yan S M,Dong G Q,Zhang L,Wang R,Liang C,Zhang R S.Anal.Test.Technol.Instrum.(吴忠平,郑水庆,严松茂,东国卿,张立,汪蓉,梁晨,张润生.分析测试技术与仪器),2012,18(4):197-203.[17] Qian Z H,Qiao H W,Hua Z D.Chin.J.ForensicMed.(钱振华,乔宏伟,花镇东.中国法医学杂志),2015,30(1):1-4.
[18] Hua Z D,Jia W,Li K.PoliceTechnol.(花镇东,贾薇,李康.警察技术),2013,(4):15-18.
[19] Xu P,Lin W S,Li X N,Liu K L,Ling X M,Lu W.Chin.J.Pharm.Anal.(徐鹏,林文斯,李晓娜,刘克林,凌笑梅,卢炜.药物分析杂志),2013,33(9):1538-1541.
[20] Xu P,Li X N,Liu K L,Ling X M,Lu W.Chin.J.ForensicMed.(徐鹏,李晓娜,刘克林,凌笑梅,卢炜.中国法医学杂志),2012,27(6):477-479.
[21] Xu P,Li X N,Liu K L,Ling X M,Lu W.Chin.J.DrugDepend(徐鹏,李晓娜,刘克林,凌笑梅,卢炜.中国药物依赖性杂志),2012,21(4):282-284.
[22] Zhai W F,Zhang C S,Gao L S.J.Instrum.Anal.(翟晚枫,张春水,高利生.分析测试学报),2014,33(8):893-898.
Simultaneous Determination of 10 Synthetic Cannabinoids in Novel “Spice” Drugs by HPLC-MS/MS
ZHANG Chun-shui,ZHAI Wan-feng*
(Institute of Forensic Science Ministry of Public Security,Beijing 100038,China)
Taking into account the high number of synthetic cannabinoids found in seized novel “spice” drugs in recent years,this study aimed at the simultaneous determination of common synthetic cannabinoids.An HPLC-MS/MS method was developed for the determination of ten synthetic cannabinoids in novel “spice” drugs.Samples were dissolved with methanol,and then ultrasound-assistedly extracted and filtered through a 0.22 μm membrane filter.The extract was separated on an Agilent Poroshell 120 EC-C18(3.0 mm×50 mm,2.7 μm) column at 30 ℃,using methanol-water as mobile phase.The flow rate was set at 0.3 mL/min.ESI+and ESI-mode were used at different times,and the MS spectra characteristics and proposed fragmentation of 10 synthetic cannabinoids were studied.Under the optimized conditions,good linear relationships were obtained in the ranges of 1-100 ng/mL for 7 synthetic cannabinoids under ESI+mode,and 10-1 000 ng/mL for 3 synthetic cannabinoids under ESI-mode.The intra-day relative standard deviations(RSDs) were not more than 3.2% and the inter-day RSDs were not more than 6.3%.This method was applied in real cases,and was proved to be fast,accurate,sensitive and precise for the determination of those 10 common synthetic cannabinoids in novel “spice” drugs.
high performance liquid chromatography tandem mass spectrometry(HPLC-MS/MS); spice; drugs;synthetic cannabinoids
2015-07-30;
2015-09-01
国家科技专项项目(2012YQ12004909)
10.3969/j.issn.1004-4957.2016.03.002
O657.63; TQ251.34
A
1004-4957(2016)03-0264-07
*通讯作者:翟晚枫,研究实习员,研究方向:毒物毒品分析检验,Tel: 010-63434750,E-mail: 765838505@qq.com
猜你喜欢
小学科学(学生版)(2021年9期)2021-11-02
山东第一医科大学(山东省医学科学院)学报(2021年7期)2021-10-13
中成药(2021年5期)2021-07-21
昆明医科大学学报(2021年2期)2021-03-29
昆明医科大学学报(2020年12期)2021-01-26
——香草传
动漫星空(兴趣百科)(2020年11期)2020-11-09
疯狂英语·新阅版(2019年10期)2019-09-10
山东化工(2019年11期)2019-06-26
国际呼吸杂志(2019年1期)2019-03-08
儿童故事画报·发现号趣味百科(2017年1期)2017-06-01
