
NOTCH3基因测序发现一个新的致病突变CADASIL诊断的复杂性
2016-12-19 07:58姬晓昙王迪龙范玉华
中风与神经疾病杂志 2016年6期
林 晶, 姬晓昙, 王迪龙, 范玉华
NOTCH3基因测序发现一个新的致病突变CADASIL诊断的复杂性
林 晶, 姬晓昙, 王迪龙, 范玉华
目的 通过一个新发现的伴皮质下梗死和白质脑病的常染色体显性遗传性脑动脉病(cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy,CADASIL)致病基因,说明基因测序对于CADASIL诊断的必要性及CADASIL诊断的复杂性。方法 对1例疑似CADASIL患者的临床表现、实验室检查、影像学检查、家系及基因测序进行分析。临床上主要表现为反复缺血性卒中及进行性记忆力下降;实验室检查排除一些高凝疾病及血管炎;头部MRI提示皮质下白质广泛对侧性信号异常及多部位的新旧梗死病灶;家系分析患者家族成员存在偏头痛、反复卒中及记忆力下降;基因测序提示NOTCH3基因3号外显子存在一个基因突变(c.331G>T p.Gly111Cys),而这个突变位点截止到目前并未报道过。结果 结合患者临床表现、影像学、家系及基因测序分析确诊该患者为CADASIL,通过分析CADASIL突变基因的致病机制确认该患者存在的突变基因为CADASIL一个新发现的致病突变。结论 新的CADASIL致病基因突变的发现强调了基因测序及提高对突变基因致病机制认识的重要性。
CADASIL; NOTCH3; 新的突变; 基因测序; 外显子
CADASIL是一种遗传性脑小血管病,临床上主要表现为反复的脑缺血发作、先兆偏头痛、认知功能障碍等异常。影像学以MRI T2像与Flair出现颞极和外囊的脑白质高信号为主要特点,侧脑室旁斑点状脑白质疏松逐步进展为广泛的脑白质病变是诊断CADASIL的有力补充[1]。
CADASIL的致病基因是位于19号染色体短臂的NOTCH3,该基因有33个外显子编码,是由2321个氨基酸构成的单次跨膜异二聚体受体蛋白Notch3,Notch3主要包括3个区域:胞外域(Notch3 extracellular domain,N3ECD)主要由34个表皮生长因子样重复序列(epidermal growth factor-like repeat,EGFr)构成、跨膜域、胞内域;其中每个EGFr包含6个半胱氨酸残基。目前有超过230种不同的致病突变报道,大部分集中在NOTCH3的2-24号外显子,其中3号外显子及4号外显子是突变热点。
临床表现及影像学可以为诊断CADASIL提供初步的参考证据,但CADASIL的诊断金标准是基因检测,尽管CADASIL被证明是脑小血管病,但微小动脉内膜平滑肌退行性变及颗粒状嗜锇样物质(granular osmiophilic material,GOM)沉积是系统性改变,皮肤小血管也存在同样病变,当基因检测发现未曾报道的突变基因时,皮肤活检可以作为重要的协诊依据。
目前针对CADASIL无特异性治疗,两种基因治疗-使用抑制性RNA分子使突变等位基因沉默或抗转录治疗介导的NOTCH3包含缺陷序列的外显子跳跃将有望成为CADASIL特异性有效治疗;现在对CADASIL只能对症治疗,例如节制吸烟、传统止痛药治疗偏头痛;抗凝药物及抗血小板药物已经证明无明确疗效,却会增加出血风险;关于认知功能障碍、胆碱酯酶抑制剂也许有效,有报道多奈哌齐可以适度改善执行功能但是机制并清楚[2]。
1 方法与结果
患者,男性,37岁。主要以突发左侧肢体无力及进行性记忆力下降入中山大学附属第一医院神经科,患者有6 y前突发言语不清、饮水呛咳及1 y前突发左侧肢体麻木病史,既往无记忆力或计算力下降、失语等认知功能障碍、情感障碍及偏头痛等病史,无高血压、糖尿病、房颤等病史,有大量吸烟史14 y;体查可发现左侧肢体上运动神经元损害(左侧肢体肌力4级、肌张力增高、腱反射活跃,病理征阳性)及高级神经活动-短时及长时记忆均有轻度下降,MMSE评分为28/30(定向力、注意力及计算力、语言能力均正常)。患者家系分析:患者母亲(已死亡)约40岁时开始出现偏头痛、反复出现脑缺血发作及进行性记忆力下降;患者姐姐现41岁,38岁开始出现偏头痛及进行性记忆力下降,未出现脑缺血发作;其他亲属未出现偏头痛、脑缺血发作及认知功能减退(见图1)。
血常规、肝肾功能、电解质、血糖、血脂及同型半胱氨酸均未见异常,自身免疫指标包括ANA、抗-dsDNA、ANCA、抗心磷脂组合、狼疮抗体等均阴性;头部MRI示:双侧基底节、放射冠、左侧丘脑、延髓右侧多发新旧不同的梗死灶,皮质下白质广泛对侧性信号异常,符合CADASIL的头部影像学改变(见图2)。对CADASIL病NOTCH3基因突变热区(3-6/11-14/18-19)进行测序,结果显示:NOTCH3 c.331G>T 3号外显子 p.(Gly111Cys)杂合;非 SNP(见图3)。NOTCH3基因相关的数据库,HGMD数据库(Human Gene Mutation Database)显示到2015年3月份更新的数据为止,没有查到c.331G>T突变的报道,最相近的是c.328的报道(见图4)。
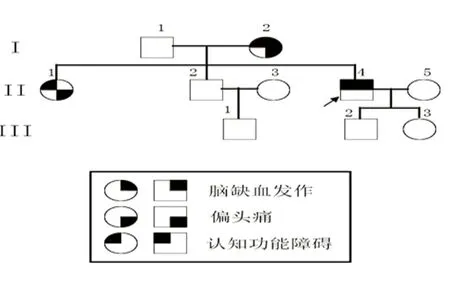
图1 该CADASIL患者的家系图谱:箭头代表先证者,正方形代表男性,圆形代表女性
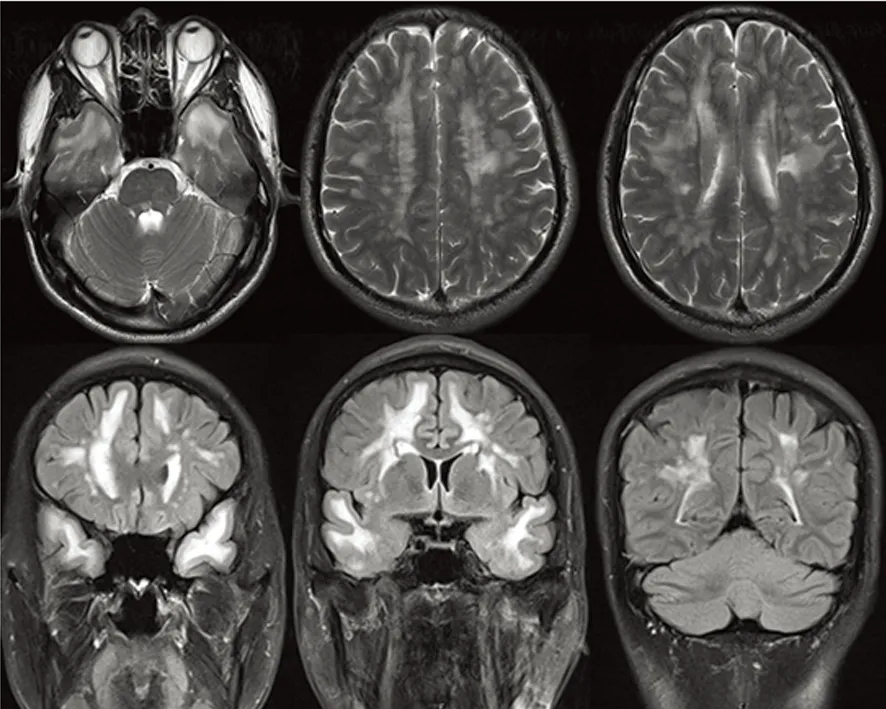
图2 头部MRI T2像及Flair示:双侧颞极、皮质下白质广泛对称性高信号,符合CADASIL影像学改变
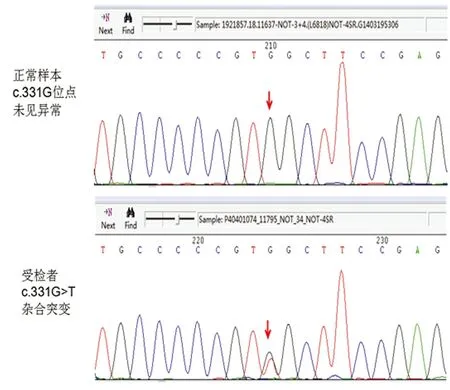
图3 CADASIL致病基因NOTCH3突变热区(3-6/11-14/18-19)测序发现-NOTCH3 c.331G>T 3号外显子
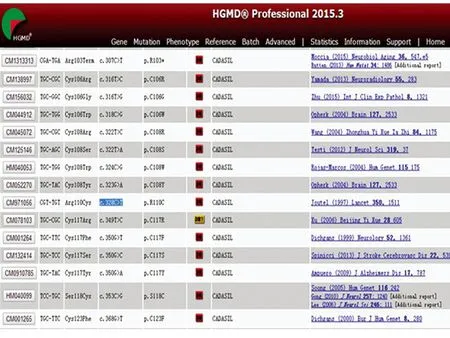
图4 NOTCH3的基因相关的数据库,HGMD数据库(Human Gene Mutation Database)显示到2015年3月份更新的数据为止,没有查到c.331G>T突变的报道,最相近的是c.328的报道
2 讨 论
CADASIL临床症状存在多样性,在不同的家系临床症状常常不同,可能是由于不同的致病突变引起[3],即使在同一个家系,不同的家系成员临床症状也有不同,具体原因不清。部分患者存在基因突变且MRI上存在明显的病灶却没有任何症状[4,5],然而有的患者只表现为一种典型的临床症状,例如先兆偏头痛、脑缺血发作或认知功能障碍[6,7]。约1/4的患者会出现偏头痛,常常在20~40岁期间起病;脑缺血发作在60%~80%的患者出现,一般会出现2~3次的急性发作,起病年龄主要集中在30~50岁之间;认知功能障碍是进行性进展,60~70岁左右可能进展为痴呆;情感障碍主要是抑郁,发病率约占CADASIL患者的10%~20%,在任何年龄都会发病,主要集中在40~50岁。
CADASIL的影像有相应的特点,MRI T2像与Flair常常出现对称性脑白质高信号,尤以颞极及外囊明显,出现类似影像学改变还需与其他疾病鉴别例如伴皮质下梗死和白质脑病的常染色体隐性遗传性脑动脉病(cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy,CARASIL)、多发性硬化(multiple sclerosis,MS)、皮质下动脉硬化性脑病(Binswanger病)等。CARASIL与CADASIL的临床症状和影像学十分相似,鉴别两者的主要方法是基因诊断及皮肤活检,基因检测是金标准,CADASIL突变基因是NOTCH3基因,而CARASIL突变基因是HTRA1基因,而皮肤活检检测是否有GOM沉积也是协助区分CARASIL与CADASIL的重要方法;MS常常累及脊髓与视神经,头部影像学很少出现对称性颞极病灶,而以垂直于侧脑室的散在病灶常见,脑脊液寡克隆带阳性也可协助诊断;Binswanger病常常有高血压病史,影像学可见脑室周围白质弥散性损害,一般没有颞极对称性损害。
该患者分别于31岁及37岁出现过两次急性卒中,目前开始出现轻度记忆力下降,从未出现偏头痛,家系分析证实患者母亲及姐姐都出现了偏头痛及记忆力下降的临床表现,母亲还存在反复脑缺血发作,影像学MRI T2像及Flair的改变符合CADASIL影像学特点,结合患者临床表现及影像学改变提示CADASIL的诊断。
CADASIL的确诊是非常复杂的,组织病理学特点主要是脑白质区域微小动脉管壁增厚、内膜PAS阳性染色、微小动脉GOM沉积,由于无法对患者深部脑白质进行活检,所以通过脑组织活检来确诊CADASIL是极其困难的;CADASIL是系统性改变,通过皮肤活检检测微小动脉GOM沉积可以作为一种辅助诊断方法,然而皮肤微小动脉GOM沉积对于诊断CADASIL特异性很高但并不十分敏感[8,9],所以皮肤活检也存在相应的局限性。
当基因检测发现一个新的突变,判断是否为CADASIL的致病突变对于确诊CADASIL是极其重要的;大部分的NOTCH3基因突变都是错义突变,错义突变导致半胱氨酸残基数量不配对从而引起N3ECD结构改变及毒性增加被认为是CADASIL经典的致病机制,但是其他类型例如基因内小缺失、复制及接合部位的突变都曾报道过,其中缺失有时不会引起半胱氨酸残基数量的改变,而是通过改变半胱氨酸残基之间的距离从而影响二硫键形成而致病。同时也有报道没有引起半胱氨酸数量改变的错义突变依然可认为是致病突变,已经发现p.Ala1020Pro,p.Arg213Lys,p.Tyr1098Ser and p.Arg75Pro的改变通过皮肤活检协诊为CADASIL,它们的氨基酸替换并没有涉及半胱氨酸,所以没有非常充分的证据表示不涉及半胱氨酸的错义突变和CADASIL发病无关[10~13]。目前有超过230种不同的致病突变报道,致病突变常常在外显子2-24号,2-24号外显子主要编码EGFr,2-24号外显子又以3号和4号外显子的突变最为常见;突变的外显子因国家不同流行程度也有所不同,在法国、德国及英国,最常见的是4号外显子,其次是3号外显子[14~16],然而在荷兰,CADASIL致病外显子最常见的是4号外显子,其次却是11号外显子[17];在中国大陆,NOTCH3突变位点主要集中在4号外显子及3号外显子,Liu等对中国大陆57个家族62个患者进行基因分析发现突变位点大部分集中在4号外显子59.6%(34/57)及3号外显子22.8%(13/57),其中碱基替换以4号外显子c.505C>T(9/57)最常见,其次是c.544C>T(7/57)[18]。然而在中华台北突变主要集中在11号外显子,Liao等对中华台北95个家族112个患者进行基因测序得到20个不同的基因突变且都是错义突变导致的半胱氨酸不配对,在11号外显子(c.1630C>T)是最常见的突变,占70.5%(67/95),少于1/5的突变集中在3-6号外显子[19]。
该患者基因检测结果显示NOTCH3基因331位(位于3号外显子)T替换了G,从而导致翻译蛋白第11位氨基酸残基由甘氨酸(Gly)变成了半胱氨酸(Cys),虽然这种突变为首次报道,是一个新的基因突变,但NOTCH3基因331位是位于3号外显子,而3号外显子是突变最为常见的部位之一,重要的是该突变引起甘氨酸变成半胱氨酸,增加了一个半胱氨酸,NOTCH3基因突变造成半胱氨酸数量的不配对是CADASIL经典的致病机制,故认为此基因突变为CADASIL致病突变。
3 结 论
虽然这个新的基因突变-NOTCH3基因331位(位于3号外显子)T替换了G导致翻译蛋白中第11位氨基酸残基由甘氨酸(Gly)变成了半胱氨酸(Cys)未曾有过报道,但是这个突变位点位于CADASIL致病基因NOTCH3突变热区,且该突变导致的半胱氨酸数量不配对是符合CADASIL普遍认可的致病机制,同时结合患者临床表现、影像学及家系分析均支持该新的基因突变是CADASIL的致病突变。
这个新的致病基因的发现同时说明基因检测对于CADASIL的诊断及寻找新的致病基因是非常有效的工具,基因检测的发展将对我们了解CADASIL及未来治疗CADASIL起着关键的作用。
[1]Salomon N.Neuroimaging of small vessel disease[J].Brain Pathol,2014,24(5):519-524.
[2]Dichgans M,Markus HS,Salloway S,et al.Donepezil in patients with subcorticalvascular cognitive impairment:a randomised double-blind trial in CADASIL[J].Lancet Neurol,2008,7(4):310-318.
[3]Joutel A,Monet M,Domenga V,et al.Pathogenic mutations associated with cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy differently affect Jagged1 binding and Notch3 activity via the RBP/JK signaling pathway[J].Am J Hum Genet,2004,74(2):338-347.
[4]Mourad A,Levasseur M,Bousser MG,et al.CADASIL with minimal symptoms after 60 years[J].Rev Neurol,2006,162(8~9):827-831.
[5]Pescini F,Bianchi S,Salvadori E,et al.A pathogenic mutation on exon 21 of the NOTCH3 gene causing CADASIL in an octogenarian paucisymptomatic patient[J].Neurol Sci,2008,267(1~2):170-173.
[6]Chabriat H,Levy C,Taillia H,et al.Patterns of MRI lesions in CADASIL[J].Neurology,1998,51(2):452-457.
[7]Pradotto L,Azan G,Doriguzzi C,et al.Sporadic vascular dementia as clinical presentation of a new missense mutation within exon 7 of NOTCH3 gene[J].Neurol Sci,2008,271(1~2):207-210.
[8]Markus HS,Martin RJ,Simpson MA,et al.Diagnostic strategies in CADASIL[J].Neurology,2002,59(8):1134-1138.
[9]Dotti MT,Federico A,Mazzei R,et al.The spectrum of Notch3 mutations in 28 Italian CADASIL families[J].Neurol Neurosurg Psychiatry,2005,769(5):736-738.
[10]Wang Z,Yuan Y,Zhang W,et al.NOTCH3 mutations and clinical features in 33 mainland Chinese families with CADASIL[J].J Neurol Neurosurg Psychiatry,2011,82(5):534-539.
[11]Mizuno T,Muranishi M,Torugun T,et al.Two Japanese CADASIL families exhibiting Notch3 mutation R75P not involving cysteine residue[J].Intern Med,2008,47(23):2067-2072.
[12]Kotorii S,Takahashi K,Kamimura K,et al.Mutations of the notch3 gene in non-Caucasian patients with suspected CADASIL syndrome[J].Dement Geriatr Cogn Disord,2001,12(3):185-193.
[13]Scheid R,Heinritz W,Leyhe T,et al.Cysteine-sparing notch3 mutations:cadasil or cadasil variants[J].Neurology,2008,71(10):774-776.
[14]Joutel A,Vahedi K,Corpechot C,et al.Strong clustering and stereotyped nature of Notch3 mutations in CADASIL patients[J].Lancet,1997,350(9090):1511-1515.
[15]Markus HS,Martin RJ,Simpson MA,et al.Diagnostic strategies in CADASIL[J].Neurology,2002,59(8):1134-1138.
[16]Peters N,Opherk C,Bergmann T,et al.Spectrum of mutations in biopsy-proven CADASIL:implications for diagnostic strategies[J].Arch Neurol,2005,62(7):1091-1094.
[17]Oberstein SA.Diagnostic strategies in CADASIL[J].Neurology,2003,60(12):2020.
[18]Liu X,Y Zuo,Sun W,et al.The genetic spectrum and the evaluation of CADASIL screening scale in Chinese patients with NOTCH3 mutations[J].Journal of the Neurological Sciences,2015,354 (1~2):63-69.
[19]Liao Y,Hsiao C,Fuh J,et al.Characterization of CADASIL among the Han Chinese in Taiwan:distinct genotypic and phenotypic profiles[J].PloS ONE,2015,10(8):e0136501.
Sequence analysis of the NOTCH3 gene discover a novel pathogenic mutation:complexity in the diagnosis of CADASIL
LIN Jing,JI Xiaotan,WANG Dilong,et al.
(Department of Neurology,First Affiliated Hospital of Sun Yat-Sen University,Guangzhou 510080,China)
Objective To emphasis the the importance of genetic tests and the complexity in the diagnosis of cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) by discovering a novel mutation.Methods Analyze clinical manifestation,laboratory examination,imaging examination,pedigree investigation and genetic tests of a patient suspected CADASIL.The clinical manifestation included recurrent ischemic stroke and progressive memory loss.Laboratory tests excluded hypercoagulable disorders and vasculitides.Magnetic resonance imaging (MRI) of the patient’s brain revealed symmetric and extensive abnormal signal in the subcortical white matter and novel or old infarction lesions.The family member of the patient had the history of migraine,recurrent stroke and memory loss.Sequence analysis of hot point mutations of the NOTCH3 gene,unveiled a novel mutation,c.331G>T (p.Gly111Cys 3 exon),which have not detected in Human Gene Mutation Database up to now.Results Combined with the characteristics of clinical manifestation,imaging of brain,family history and genetic sequence,the patient was identified as CADASIL and the novel mutation had been considered as the pathogenic mutation of CADASIL.Conclusion The fingding of the novel pathogenic gene emphasize the significance of genetic tests and the cognition in pathogenesis of mutation gene.
CADASIL; NOTCH3; Novel mutation; Genetic sequence; Exon
1003-2754(2016)06-0492-04
2016-02-09;
2016-03-29
广东省省科技计划项目(2015A030302013)
(中山大学附属第一医院神经科,广东 广州 510080)
范玉华,E-mail:fansusan@126.com
R742
A
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
昆明医科大学学报(2021年3期)2021-07-22
中国生殖健康(2020年4期)2021-01-18
医学新知(2019年4期)2020-01-02
中国生殖健康(2018年4期)2018-11-06
中华老年多器官疾病杂志(2016年8期)2016-05-14
湖北农业科学(2014年11期)2014-09-10
中医研究(2014年6期)2014-03-11
中医研究(2014年4期)2014-03-11
中国神经精神疾病杂志(2014年1期)2014-03-01
