
高效液相色谱-串联质谱法同时测定供港生猪尿液中29种限用兽药残留
2016-12-01 01:12伍华雯张远芳万建春占春瑞
分析测试学报 2016年10期
伍华雯,郭 平,张远芳,曾 赞,万建春,占春瑞
(1.江西出入境检验检疫局 综合技术中心,江西 南昌 330035;2.东北林业大学野生动物资源学院,黑龙江 哈尔滨 150040)
高效液相色谱-串联质谱法同时测定供港生猪尿液中29种限用兽药残留
伍华雯1*,郭 平1,张远芳1,曾 赞2,万建春1,占春瑞1
(1.江西出入境检验检疫局 综合技术中心,江西 南昌 330035;2.东北林业大学野生动物资源学院,黑龙江 哈尔滨 150040)
建立了高效液相色谱-串联质谱(HPLC-MS/MS)同时测定生猪尿液中喹诺酮类、磺胺类、磺胺增效剂、四环素类、林可胺类、大环内脂共29种限用兽药残留量的检测方法。试样经乙酸铵和EDTA-Na缓冲液提取,HLB固相萃取小柱净化后,HPLC-MS/MS进行测定,其中β-受体激动剂类用内标法定量,其余兽药用外标法定量。在电喷雾电离正离子模式下,以多反应监测(MRM)方式采集数据进行定性与定量分析。29种兽药在猪尿基质中标准曲线的线性系数(r)均大于0.99,3个不同加标水平下的平均回收率为58%~108%,日内相对标准偏差(RSD)为1.9%~18.9%,日间RSD为3.4%~20.9%;定量下限(LOQ,S/N≥10)为1.0~10.0 μg/L。该方法经济、高效、可靠,可用于生猪屠宰前兽药多残留的快速检测。
高效液相色谱-串联质谱法(HPLC-MS/MS);生猪尿液;限用兽药;多残留
随着赣港经贸的快速发展,江西对港出口呈连续增长趋势,供港生猪尤为明显,目前年供应量已居全国前列,香港市场约30%猪肉来自江西。香港对进口农副产品有严格质量要求,《食物内有害物质规例》中规定了生猪的46种兽药限量标准,在一定程度上给江西供港生猪企业带来障碍。目前,生猪体内兽药残留监控主要通过尿液检测来实现,但我国猪尿中兽药多残留检测标准严重缺乏,参考方法存在通量低的不足,难以满足生猪供港快速通关要求。
目前采用液相色谱法、高效液相色谱法和高效液相色谱-串联质谱法测定动物源性食品中兽药残留已有较多报道[1-5]。而在兽药残留检测基质趋于多样化的当下,猪尿中兽药残留的研究多为β-受体激动剂方面的检测[6]。虽有同时测定多种兽药残留的方法报道[7-16],但同时对猪尿中喹诺酮类、磺胺类、磺胺增效剂、林可胺类、大环内酯类5大类药物残留检测技术的研究尚未见报道。本研究采用高效液相色谱-串联质谱法同时检测生猪尿液中29种限用药物的残留量,以期实现生猪屠宰前兽药多残留的快速检测。
1 实验部分
1.1 仪器与试剂
API4000+三重四极杆液相色谱-串联质谱仪(美国AB Sciex公司),配30A系列高效液相色谱仪(日本岛津公司);电子天平(德国Sartorius公司,感量0.01 g和0.1 mg);旋涡混合器(德国IKA公司);Eppendorf 5430R 冷冻高速离心机(德国Eppendorf公司);N-EVAP 112氮吹浓缩仪(美国Organomatian Associates公司);Visiperp DL固相萃取装置(美国Supelco公司);KQ-600B型超声波清洗器(昆山市超声仪器有限公司);SI G560E型旋涡混合器(美国Scientific Industries公司);pH计(美国Mettler Toledo公司);Waters ACQUITY UPLC BEH RP C18色谱柱(100 mm×2.1 mm,粒径1.7 μm,美国Waters公司)。
生猪尿样采自养殖场;阴性猪尿样品采自养殖场中未给药的生猪,经实验室处理后上机测定,所得谱图与标准溶液的谱图进行比对加以确证。
甲醇、乙腈(HPLC纯,德国Merck公司);甲酸(HPLC 纯,上海安普公司);甲酸铵(HPLC 纯,瑞士Sigma-Aldrich 公司);无水乙酸铵(分析纯,国药集团化学试剂有限公司);氨水、乙二胺四乙酸钠(分析纯,广州西陇化工化学试剂有限公司);HLB固相萃取小柱(500 mg,6 mL,美国Waters公司);Milli-Q超纯水(GB/T6682 规定的一级水)。
标准物质:29种兽药标准物质的纯度在93.0%~99.0%范围内,均购于德国Dr.Ehrenstorfer公司。
标准溶液:1.0 g/L,取适量上述29种兽药标准物质,分别用甲醇溶解配制成标准储备溶液,-18 ℃保存于棕色玻璃瓶中。使用前以甲醇稀释成合适浓度的标准工作液。
提取液:称取15.4 g乙酸铵,加水溶解并定容至1 L,混匀,用乙酸调至pH 5.2,得到0.2 mol/L乙酸铵溶液;称取37.2 g乙二胺四乙酸二钠,加水溶解并定容至1 L,混匀后即得0.1 mol/L EDTA-2Na。
流动相:准确称取0.315 3 g甲酸铵,加800 mL水溶解,准确加入1 mL甲酸,用水定容至1 L,摇匀,得到5 mmol/L甲酸铵(含0.1%甲酸)。
溶解液:乙腈-流动相(2∶8,体积比)。
1.2 样品预处理
1.2.1 样品提取及净化 准确移取(2±0.05) mL试样,置于50 mL具塞聚丙烯离心管中,加6 mL 0.2 mol/L乙酸铵缓冲溶液(乙酸调至pH 5.2)和6 mL 0.1 mol/L EDTA溶液,涡旋,4 500 r/min离心5 min。
上清液转入HLB固相萃取柱(预先经5 mL甲醇、5 mL水活化),依次用5 mL水、5 mL 0.5%的氨水、5 mL甲醇水(1∶9)淋洗小柱,真空抽干小柱,再依次用5 mL甲醇、5 mL浓氨水-甲醇(5∶95)洗脱,洗脱液于40 ℃下氮气吹干,准确加入1 mL溶解液复溶,超声,涡旋均匀,过0.22 μm滤膜,供液相色谱-串联质谱仪分析。
1.2.2 空白基质溶液的制备 准确移取(2±0.05) mL阴性猪尿试样,按“1.2.1”操作步骤进行处理。
1.3 色谱-质谱条件
1.3.1 色谱条件 柱温45 ℃;流速200 μL/min;进样体积5 μL;流动相:A为5 mmol/L甲酸铵(含0.1%的甲酸),B为乙腈。梯度洗脱程序:0~2 min,20%B;2~3 min,线性递增至24%B;3~5 min线性递增至28%B;5~5.5 min,保持28%B;5.5~7 min,线性递增至32%B;7~10.5 min,线性递增至36%B;10.5~12 min,线性递增至95%B;12~12.01 min,线性递减至5%B;12.01~15 min,5%B。
1.3.2 质谱条件 ESI正离子模式扫描(ESI+),多反应监测模式(MRM),雾化气压力(GSI):60 psi;辅助气压力:60 psi;气帘气压力(CUR):35 psi;离子源温度:600 ℃;电喷雾压力(IS):5 500 V;单位分辨率扫描;碰撞气体流速:中流速。
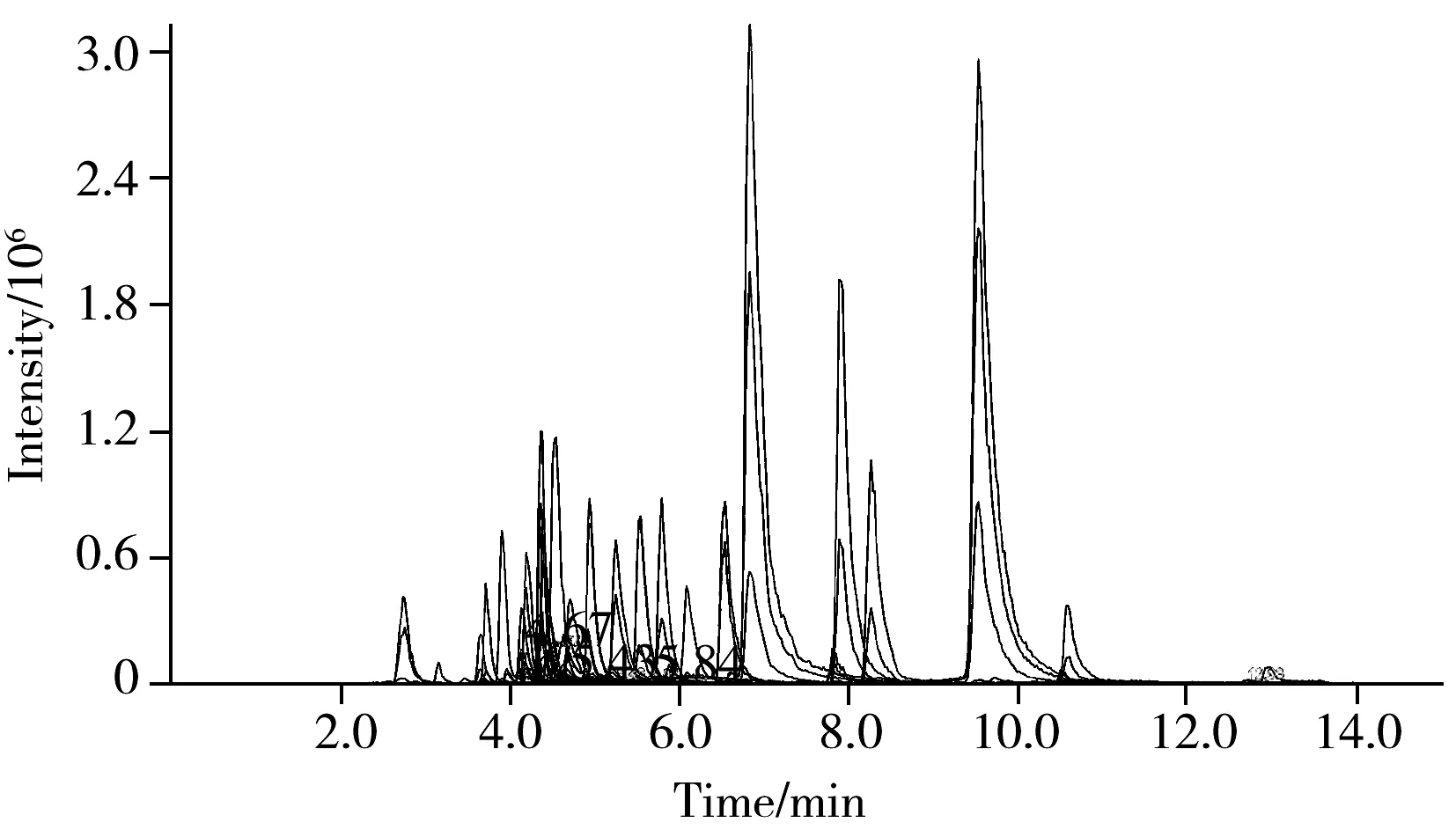
图1 29种化合物混合标准溶液的MRM色谱图Fig.1 MRM chromatogram of 29 analytes mixed standard solution
2 结果与讨论
2.1 色谱-质谱条件的优化
2.1.1 色谱条件的选择 因待测的化合物种类较多,采用普通C18色谱柱分离所需时间较长,故本实验采用超高效液相色谱柱Waters ACQUITY UPLCBEH C18(2.1 mm×100 mm,1.7 μm)进行分离。采用5 mmol/L甲酸铵(含0.1%甲酸)-乙腈作为流动相,通过调节流动相的洗脱梯度,各化合物的色谱峰分离良好,峰形尖锐。29种化合物的混合标准溶液色谱图如图1所示。
2.1.2 质谱条件的优化 去簇电压(DP)和碰撞气能量(CE)直接影响定性和定量离子的丰度,与方法的灵敏度密切相关[10]。本实验对0.2 mg/L的各化合物标准溶液进行全扫描,结合各化合物的理化性质,在Q1MS(Q1)模式下确定母离子,Product Ion(MS2)模式选定2个丰度较高、干扰较少的子离子分别作为定性和定量离子,并依次对各化合物子离子的去簇电压(DP)、碰撞气能量(CE)、入口电压(EP)及碰撞室出口电压(CXP)进行优化。其中EP和CXP基本在10~12 V左右,其余参数结果见表1。
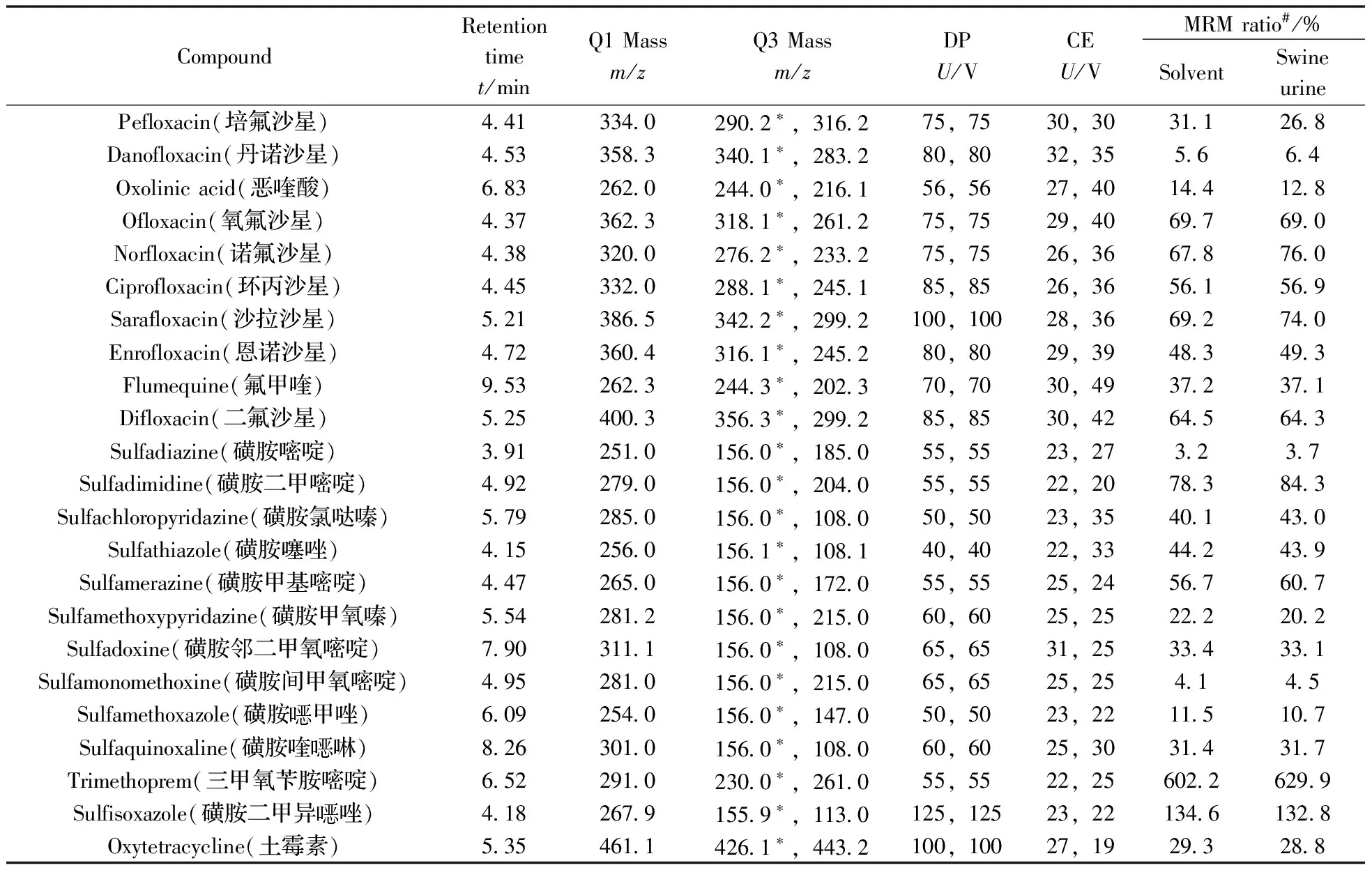
表1 29种兽药的多反应监测(MRM)质谱相关参数
(续表1)
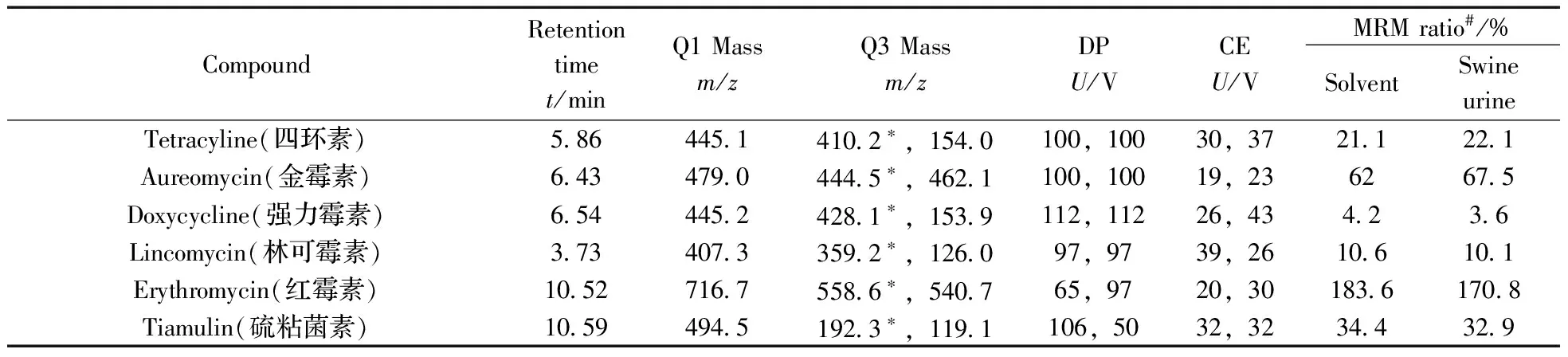
CompoundRetentiontimet/minQ1Massm/zQ3Massm/zDPU/VCEU/VMRMratio#/%SolventSwineurineTetracyline(四环素)58644514102∗,1540100,10030,37211221Aureomycin(金霉素)64347904445∗,4621100,10019,2362675Doxycycline(强力霉素)65444524281∗,1539112,11226,434236Lincomycin(林可霉素)37340733592∗,126097,9739,26106101Erythromycin(红霉素)105271675586∗,540765,9720,3018361708Tiamulin(硫粘菌素)105949451923∗,1191106,5032,32344329
*:quantitative ion(定量离子);#:average ratios of lincomycin and erythromycin at 1.0,2.0,4.0 μg/L and the other analytes at 10,20,40 μg/L(林可霉素和红霉素在1.0,2.0,4.0 μg/L,其余化合物在10,20,40 μg/L加标水平下,其相应离子相对丰度比的平均值)
2.2 样品前处理条件的优化
2.2.1 提取方法的优化 与单残留方法相比,多残留检测的提取方法显得颇为关键。喹诺酮类、磺胺类兽药均呈现酸碱两性,在酸性溶剂中更易被萃取;大环内酯类化合物在酸性条件下易开环,不稳定[13],故提取液的酸度不宜过大;四环素族的化合物易与金属离子发生螯合作用形成螯合物[14]。综上原因,本方法采用乙酸铵和乙二胺四乙酸二钠的缓冲液进行提取。
2.2.2 净化条件的优化 相比动物组织肌肉、肝脏等基质,尿样基质成分较简单,因此本实验主要结合待测化合物的理化性质进行净化条件的优化。由于待测化合物种类较多,且各化合物的极性不同,故考虑选择对酸性、碱性和中性化合物吸附范围较广的反相固相萃取小柱。在阴性试样中分别添加不同浓度水平的29种化合物混合标准溶液,对比研究了HLB柱和PLS柱的净化效果,发现两者对大多数化合物的净化效果无明显差异,但HLB柱对高浓度添加水平的磺胺类、四环素等化合物的净化效果略优,故确定选用HLB固相萃取柱进行实验。
对淋洗液的种类进行了考察。由于尿样基质中的杂质多为水溶性,先选用水淋洗,再选择5%的甲醇水溶液淋洗,发现林可胺类等部分化合物存在明显的杂质干扰。综合考虑化合物的极性及酸碱特性,增加淋洗液中甲醇的比例后,样液中仍存在杂质干扰。进一步增加氨水淋洗,并控制氨水的浓度以防待测物被淋洗下来。对不同浓度氨水溶液、不同比例甲醇水淋洗的对比研究显示,相比仅用5%的甲醇水溶液,经0.5%氨水和10%甲醇水淋洗后,磺胺类和喹诺酮类化合物的回收率明显提高,且对林可胺类化合物的除杂质效果明显。
2.3 方法学验证
2.3.1 定量下限与基质标准曲线 在阴性尿样基质中添加一定浓度的标准溶液,在优化的色谱-质谱条件下进行测定,提取定量离子的色谱图,根据色谱峰的信噪比不小于10(S/N≥10)确定29种兽药的定量下限(LOQ)为1.0~10.0 μg/L。在选定的仪器条件下,测定6个浓度水平的系列混合标准工作溶液,以29种兽药的质量浓度为横坐标(x,μg/L),峰面积为纵坐标(y)绘制标准工作曲线。结果表明,29种兽药在1.0~200.0 μg/L质量浓度范围内线性关系良好,相关系数(r)为0.990 7~0.999 7(见表2)。
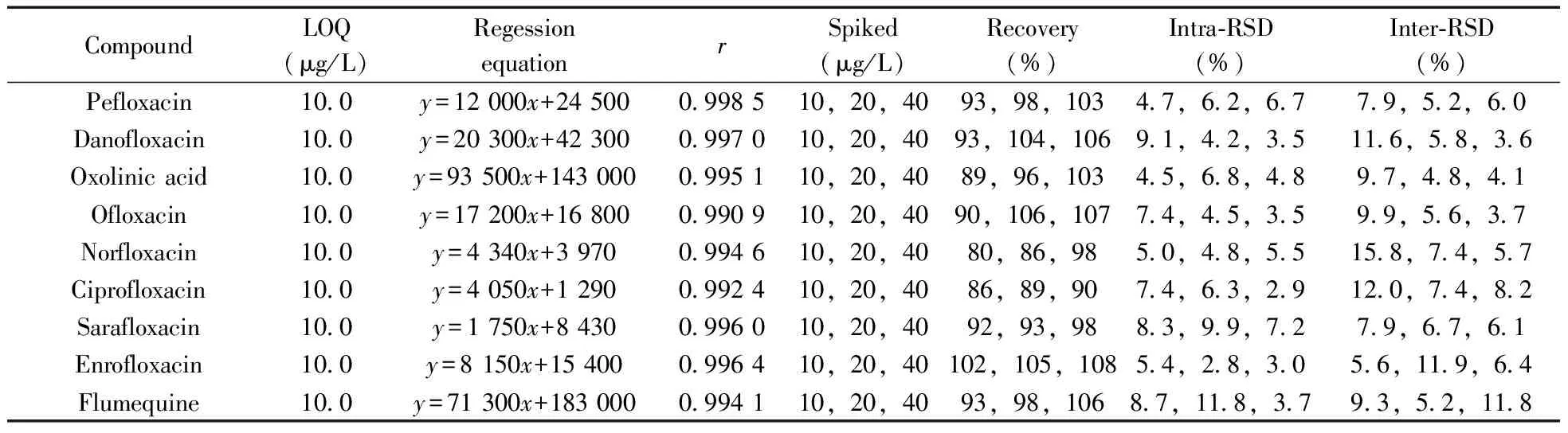
表2 29种兽药在猪尿基质中的定量下限、回归方程、相关系数(r)、回收率及相对标准偏差(n=6)
(续表2)

CompoundLOQ(μg/L)RegessionequationrSpiked(μg/L)Recovery(%)Intra⁃RSD(%)Inter⁃RSD(%)Difloxacin100y=13600x+292000995410,20,4093,98,10479,49,3369,63,36Sulfadiazine100y=9250x+83700994910,20,4070,77,9654,116,47182,123,146Sulfadimidine100y=3960x-84800997210,20,4083,72,91141,181,9947,156,104Sulfachloropyridazine100y=14500x-78900994410,20,4076,77,9586,135,43133,127,68Sulfathiazole100y=5340x+109000998310,20,4070,79,9597,157,66137,138,62Sulfamerazine100y=5320x-30600997910,20,4080,75,10079,170,53198,143,61Sulfamethoxypyridazine100y=14800x+7570994010,20,4069,74,9875,143,49195,147,49Sulfadoxine100y=43100x+1090000990710,20,4066,74,10062,179,79120,117,65Sulfamonomethoxine100y=11400x+60800998210,20,4082,78,10291,79,55209,109,51Sulfamethoxazole100y=9430x+118000994410,20,4073,80,9945,146,4072,70,63Sulfaquinoxaline100y=21700x+745000999710,20,4095,86,10679,54,38150,87,38Trimethoprem100y=932x+14600994410,20,4076,93,10568,33,68156,67,55Sulfisoxazole100y=12900x+141000997010,20,4067,72,9089,189,52198,125,103Oxytetracycline100y=3279x-91350994310,20,4095,67,7061,79,78195,203,182Tetracyline100y=4924x+21280994410,20,4067,83,8966,39,118175,108,105Aureomycin100y=1353x-35820998610,20,4080,65,58173,53,100153,34,121Doxycycline100y=9153x+22940998910,20,4078,94,10035,115,172179,149,179Lincomycin10y=51800x+531000995010,20,4099,98,10888,90,19103,96,70Erythromycin10y=13400x+65700997210,20,4095,96,91127,74,102145,78,135Tiamulin100y=8240x+1020000997410,20,4095,102,10892,32,2198,44,135
2.3.2 回收率与精密度 取阴性猪尿样液做低、中、高3个浓度水平的加标回收实验,每个水平做6个平行测定,重复3次,分别计算同一次实验同一浓度水平6个平行之间(日内)和3次重复试验之间(日间)的相对标准偏差(RSD),结果见表2。各化合物的平均回收率为58%~108%,日内RSD为1.9%~18.9%,日间RSD为3.4%~20.9%。表明该方法的回收率和重现性均较好。
2.3.3 确证分析 根据相同实验条件下,检出物质色谱峰的保留时间应与标准品一致(允许偏差为±2.5%),且扣除背景后样品谱图中各定性离子的相对丰度比与浓度接近的标准溶液谱图相比,允许偏差符合欧盟导则EC Decision 625/2002中的规定[17],可判定样品中存在对应的被测物。本实验计算了3个加标水平(林可霉素和红霉素添加1.0,2.0,4.0 μg/L,其余27种添加10,20,40 μg/L)下29种化合物在纯溶剂和猪尿基质中定性离子的相对丰度比,结果表明,29种化合物在猪尿基质中3个加标浓度水平的平均离子相对丰度比与纯溶剂中离子相对丰度比基本相当,均在欧盟导则所允许的范围内,详见表1。
2.4 实际样品的测定
使用该方法测定江西省供港生猪尿样共884批次,以四环素类和磺胺类药物的检出率较高,其中有437批次尿样中的四环素类药物残留超出方法定量下限,37批次尿样中的四环素类兽药超出《食物内有害物质规例》中的最大残留限量。
3 结 论
本文以供港生猪尿液为样本,采用乙酸铵和EDTA缓冲液体系酶解提取,HLB固相萃取柱净化,利用Waters ACQUITY UPLC BEH C18柱进行分离,建立了高效液相色谱-串联质谱同时测定生猪尿液中29种兽药残留的分析方法。该方法的定量下限满足《食物内有害物质规例》对生猪中46种兽药限量的规定,各项技术指标均能满足日常残留检测分析的要求,能有效为施检单位缩短检测时间、降低检测成本;同时也能为养殖户缩短生猪通关时间,从而节约存栏成本。
[1] Lin H D,Lin F,Zhang M J,Xie S X,Wu Y X,Shao L Z,Yao Y X.FoodSci.(林海丹,林峰,张美金,谢守新,吴映璇,邵琳智,姚仰勋.食品科学),2011,32(2):231-236.
[2] Qian Z Z,Su X H,Wei B J,Wu C Y.FoodSci.(钱卓真,苏秀华,魏博娟,吴成业.食品科学),2010,31(6):185-189.
[3] Guo W,Liu Y,Liu N,Wei D X.Chin.J.Anal.Chem.(郭伟,刘永,刘宁,魏东旭.分析化学),2009,37(11):1638-1644.
[4] Wang X L,Guo T,Wang S S,Zhao J,Yuan J P,Zhao R S.J.Instrum.Anal.(王晓利,郭涛,王珊珊,赵金,苑金鹏,赵汝松.分析测试学报),2015,34(1):115-119.
[5] GB/T 21317-2007.Determination of Tetracyclines Residues in Food of Animal Origin LC-MS/MS Method and HPLC Method.National Standard of the People’s Republic of China(动物源性食品中四环素类兽药残留量检测方法 液相色谱-质谱/质谱法与高效液相色谱法.中华人民共和国国家标准).
[6] Nie J R,Zhu M L,Lian J,Pan Y S,Deng X L,Hu C P.Chin.J.Chromatogr.(聂建荣,朱铭立,连槿,潘云山,邓香连,胡翠萍.色谱),2010,28(8):759-764.
[7] Wang X F,Zhao L,Zhang G K,Yang J W,Bian K,Wang Z N,He L M.Chin.J.Anal.Chem.(王旭峰,赵丽,张高奎,杨建文,卞愧,王宗楠,贺利民.分析化学),2013,41(8):1254-1258.
[8] Yan L Z,Zhao Y B,Fu Y,Song W,Liu N.J.Anal.Lab.(晏利芝,赵永彪,富玉,宋薇,刘宁.分析试验室),2011,30(1):84-86.
[9] Cao H,Chen X Z,Zhu Y,Li Z G,Wang L N,Zhang X B.J.Chin.MassSpectrom.Soc.(曹慧,陈小珍,朱岩,李祖光,汪丽娜,张晓波.质谱学报),2013,34(4):202-214.
[10] Xiong C L,Guo P,Zhan C R,Liu G B,Wan J C.J.Instrum.Anal.(熊春兰,郭平,占春瑞,刘光斌,万建春.分析测试学报),2013,32(2):193-198.
[11] Fu T P,Zhang F,Liu L,Chu X G,Xu C B.J.Instrum.Anal.(付体鹏,张峰,刘力,储晓刚,许成保.分析测试学报),2013,32(10):1153-1159.
[12] Shi A.Chin.J.Chromatogr.(石奥.色谱),2016,34(2):176-183.
[13] Sun L,Zhang L,Wang S H,Wang X.J.Instrum.Anal.(孙雷,张骊,王树槐,汪霞.分析测试学报),2009,28(9):1058-1061.
[14] Wang S,Zhang J,Shao B.J.Instrum.Anal.(王硕,张晶,邵兵.分析测试学报),2013,32(2):179-185.
[15] Guo L M,Zhu K,Jiang H Y,Li J C,Li X W,Ding S Y.J.Instrum.Anal.(郭黎明,朱奎,江海洋,李建成,李晓薇,丁双阳.色谱),2009,27(4):412-416.
[16] Guo D H,Deng X J,Zhao S Z,Zhu J,Xia C F,Chen S S,Song Y.Chin.J.Anal.Chem.(郭德华,邓晓军,赵善贞,朱坚,夏崇菲,陈舜胜,宋越.分析化学),2010,3(38):318-324.
[17] EC Decision 657/2002.Off.J.Eur.Commun.,2002,1221:8-36.
Simultaneous Determination of 29 Restricted Veterinary Drug Residues in Urine of Swine Supplied to Hong Kong by High Performance Liquid Chromatography-Tandem Mass Spectrometry
WU Hua-wen1*,GUO Ping1,ZHANG Yuan-fang1,ZENG Zan2,WAN Jian-chun1,ZHAN Chun-rui1
(1.Comprehensive Technology Center,Jiangxi Entry-Exit Inspection and Quarantine Bureau,Nanchang 330035,China;2.College of Wildlife Resoureces,Northeast Forestry University,Harbin 150040,China)
A new approach was established for the simultaneous determination of 29 restricted drugs,including quinolones,sulfoamides,trimethoprim,tetracyclines,lincosamides,macrolides in urine of swine supplied to Hong Kong by high performance liquid chromatography-tandem mass spectrometry(HPLC-MS/MS).The analytes were extracted with ammonium acetate and EDTA buffer,and followed by cleanup using an HLB solid phase extraction cartridge.The sample matrix-matched calibration was used to determine the residue.Electrospray ionization mass spectrometry was operated in the positive mode. The analysis of 29 restricted drugs was achieved under the multiple reaction monitoring(MRM) mode.The results indicated that correlation coefficients of the calibration curves were more than 0.99.The average recoveries of 29 analytes at three spiked concentration levels ranged from 58% to 108%.The intra-day relative standard deviations(RSDs) were in the range of 1.9%-18.9%,and the inter-day RSDs were 3.4%-20.9%.The limits of quantitations(LOQs,S/N≥10) were in the range of 1.0-10.0 μg/L.The method is economic,high efficient and reliable,and is suitable for the simultaneous determination of multiple residues of veterinary drugs in swine urine.
high performance liquid chromatography-tandem mass spectrometry(HPLC-MS/MS);swine urine;restricted drug;multiple residues
2016-04-01;
2016-05-04
江西省科技计划项目(20151BBF60038)
10.3969/j.issn.1004-4957.2016.10.007
O657.63;TQ460.72
A
1004-4957(2016)10-1261-06
*通讯作者:伍华雯,助理工程师,研究方向:食品安全与检测分析,Tel:0791-86358771,E-mail:dcwhw@163.com
猜你喜欢
今日畜牧兽医(2022年10期)2022-12-23
家庭科学·新健康(2022年2期)2022-03-07
中国蜂业(2018年4期)2018-05-09
河南畜牧兽医(2016年24期)2016-11-29
环球时报(2016-05-18)2016-05-18
当代化工研究(2016年6期)2016-03-20
兽医导刊(2015年7期)2016-01-04
色谱(2015年6期)2015-12-26
中国环境科学(2015年7期)2015-01-28
云南畜牧兽医(2014年4期)2014-02-28
