
Tau蛋白的磷酸化对阿尔兹海默症影响的相关研究
2016-11-30 06:37刘晏伊李志鹏
中国老年保健医学 2016年5期
刘晏伊 陈 超 李志鹏 杨 泽
※为通讯作者
Tau蛋白的磷酸化对阿尔兹海默症影响的相关研究
刘晏伊1,2陈 超2李志鹏2杨 泽1※
阿尔兹海默症(Alzheimer’s disease,AD)俗称老年痴呆,是一种较为常见的慢性进行性神经退行性疾病,主要表现为全脑性不可逆转性脑功能损害。其病因复杂,故发病机制尚不明确。现如今研究的其中一种理论认为Tau蛋白的异常聚合会和束状细丝(straight filament,SF)一同组成神经元纤维缠结(neurofibrillary tangles,NFTs),导致神经纤维内的微管变形,从而导致神经元变性甚至死亡。大量的神经元的死亡会导致AD的发生。因此,有必要摸清Tau蛋白异常磷酸化的机理来对患者进行有针对性的治疗。
阿尔兹海默症 Tau蛋白 神经纤维缠结
阿尔兹海默症作为最常见的一种老年痴呆,在1906年由Alzheimer教授发现第一位患者并命名。其初期发病表现不明显,主要表现为记忆力缓慢性退化,认知、执行功能障碍以及人格和行为改变等全面性痴呆。随着人口老龄化的严重,近年来AD已一跃成为导致老年人死亡的第四位主要原因,仅次于心脏病、癌症以及中风,并且带给患病家庭以沉重的精神及经济压力。因此,早日了解到AD的发病机制对于治疗该病具有非常重要的意义。现今的研究一共有两种主流的学说,一是细胞外的淀粉样多肽沉积,AD患者大脑因过度或减少清除β-淀粉样蛋白而具有过多神经元间淀粉样肽(Aβ).这样会形成密集的淀粉样低聚物,作为弥散性斑沉淀下来。这些斑块小神经胶质活化、细胞因子形成及补体级联反应活化导致炎症病变。炎症会促使神经炎斑形成,导致突触与神经炎损伤和细胞死亡。另外一种理论是细胞内的异常磷酸化的Tau蛋白沉积。本文主要对第二种机制进行综述。
1. Tau蛋白
1.1 Tau蛋白的结构 Tau蛋白的基因位于人体的第17号染色体长臂上。全长100kb,有16个外显子,其中11个编码Tau蛋白[1]。正常人中由于Tau蛋白mRNA的选择性剪切,会编码出6种亚型。分别由352,381,383,410,412和441个氨基酸残基组成,分子量为48~67KD。Tau蛋白的一级结构可分为4个区域,分别是N端区、C端区、Pro富集区和氨基酸残基的重复串联区。其中N端区为延伸结构域,从微管的外表面延伸出来,会与其他细胞骨架成分及细胞膜接触,以维持细胞轴突的稳定。其次,氨基酸残基的重复串联区其实是Tau蛋白的微管结合区,Tau蛋白可通过这个区域和微管的外表面紧紧相连,并与微管相互作用,促进微管的组装以及参与细胞轴突的运输。
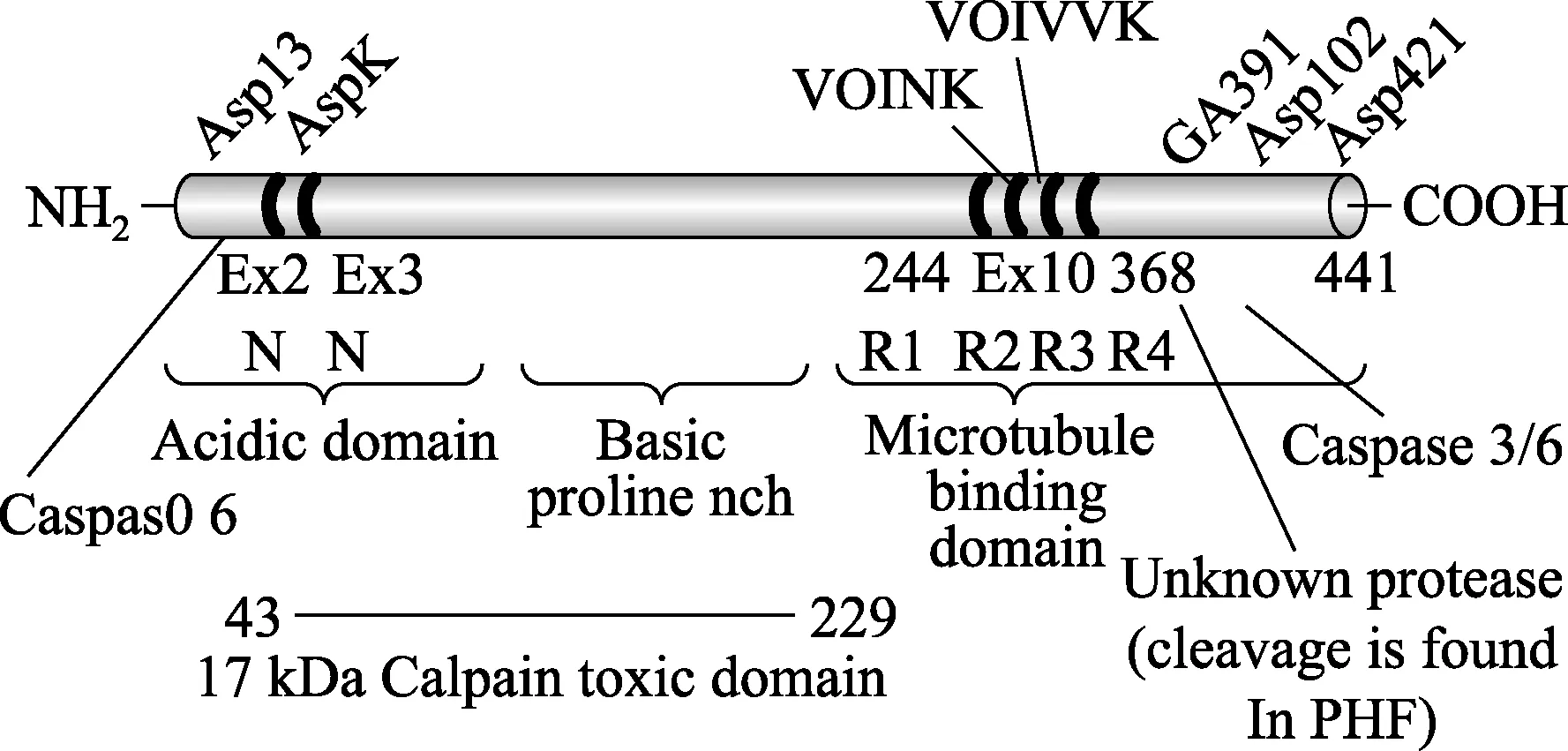
Tan蛋白示意图[2]Schematic representation of Tau Protein
1.2 Tau蛋白的特征及功能 Tau蛋白是一种结构的微管相关蛋白。一般在中枢神经系统的神经元的轴突上高度表达。通常,在正常脑中Tau蛋白会与微管蛋白结合并促进其聚合成微管,从而以维持微管的稳定性,其次可以降低微管蛋白分子的解离。它参与维持细胞形态、物质运转、细胞分裂、细胞运动及细胞内外信息传递等功能[3]。Tau蛋白的功能障碍可能导致细胞骨架失去支撑作用从而坍塌,造成神经信号的传导受损及神经物质的转运障碍,最终导致认知障碍。
可溶性Tau蛋白有2种构象:顺式的(cis)和反式的(trans)。cis-tau蛋白可以与微管结合,而trans-tau蛋白则会组装成成对螺旋纤丝(Paried helical filaments,PHFs)。磷酸化的Tau蛋白将会使其本身的构象受到影响,会让Tau蛋白维持在反式构象并稳定下来。但这种维持并不是不可改变,近年来人们发现了一种蛋白Pin1,它是一种能使trans-tau转变成cis-tau的异构酶,可以抑制PHFs的形成同时促进微管的组装。Pin1可通过抑制GSK-3β(Tau蛋白激酶Ⅰ)的活性和异化PP2A(一种活性最强的蛋白激酶酯酶)从而催化Tau蛋白去磷酸化反应以降低Tau蛋白磷酸化水平,使trans-tau蛋白转化为cis-tau,并促进其与微管结合。现在,Pin1对稳定cis-tau蛋白结构的作用被人们更多的关注。
正常人的成熟的脑中会含有2~3个磷酸基在Tau蛋白分子中。而AD患者脑中的Tau蛋白会异常过度磷酸化,从而会使其与从微管上脱落下来,这时每分子的Tau蛋白会增加至5~9个磷酸基,这使得它会丧失正常的生理功能。从微管脱落下来的Tau蛋白聚集在一起形成可溶性的Tau聚集体,随后又组装成PHFs,并最终形成NFTs变为不可溶沉淀下来使得神经元功能受损[4]。不可溶Tau蛋白组成包括PHFs的亚单位主要有过度磷酸化的Tau蛋白,和少量微管相关蛋白。Tau蛋白是组成PHFs/NFTs的唯一必需成分。在这个过程中会产生毒性。所以,可溶性的Tau聚集体和纤维状Tau蛋白会直接促成Tau蛋白介导的神经元的损伤甚至导致AD的发生。通过使用激酶抑制剂可以抑制Tau蛋白磷酸化的整个过程。Tau毒性也可以通过增强Tau的清除和降解Tau聚集体来完成。
2. Tau蛋白与AD相关病理特征之间的关系
2.1 Tau蛋白的磷酸化 Tau蛋白的磷酸化是由磷酸酶和激酶共同调节的,这种调节的失衡将导致Tau蛋白的过度磷酸化以及进一步的病理作用,如导致AD[5]。Tau蛋白磷酸激酶可以分为三类:脯氨酸蛋白激酶(proline-directed protein kinase,PDPK)、非PDPK蛋白激酶和酪氨酸蛋白激酶(tyrosine protein kinase,TPK)。Tau 蛋白磷酸化程度是由体内的多种磷酸化的特异性蛋白激酶和脱磷酸化的蛋白磷酸酯酶两种作用间互相平衡出来的结果。

Tau蛋白与磷酸化的Tau蛋白之间的相互关系
cAMP是细胞内的一种非常重要的第二信使,负责调控与G蛋白耦联手提相关的众多细胞外信号通路。EPAC是一种由cAMP可在细胞内直接激活的交换因子,参与与cAMP相关多种信号通路的调控。EPAC有EPAC1和EPAC2两种亚型,广泛分布于人体各组织与器官。现有研究证明,EPAC2的下降会导致Tau的pT205,pT231,pS396,pS404位点的磷酸化程度增加。从而激活了CDK5(细胞周期蛋白依赖性激酶,又称Tau蛋白激酶Ⅱ)。CDK5是脑组织中调节蛋白磷酸化的重要激酶,但需与激活剂p35结合才能表现出激酶活性。当细胞内游离的钙离子增加时会激活钙蛋白酶,从而将p35裂解为更稳定的p25,延长CDK5的活性[6]。
Calpain是钙离子依赖性半胱氨酸中性蛋白酶,同时也是CDK5的激活因子,参与各种细胞功能调控。当神经元发生损伤时,该稳定失调,导致Calpain过度激活,促使p35降解为p25,CDK5过度活化,从而引发神经病变性改变。有研究证实,AD患者大脑内Calpain与CDK5的含量均高于健康对照组。所以,EPAC2的下降会激活Calpain进而活化CDK5,造成Tau蛋白的异常过度磷酸化。
现今理论认为,过度磷酸化的Tau蛋白会从微管上脱落,然后聚集形成NFTs,从而导致AD。Tau蛋白的异常过度磷酸化将影响其与微管的结合能力。更为严重的是它可促进和增强不溶性Tau蛋白的聚集。
Tau蛋白的异常高度磷酸化是出现NFTs和PHFs以及束状纤丝(straight filament,SF)的主要原因,所以AD的产生与异常高度磷酸化的Tau蛋白的关系是密不可分的。
2.2 Tau蛋白与Aβ蛋白的关系 异常的Aβ蛋白可以构成有神经毒性的老年斑。正常情况下Aβ蛋白是可溶性的,但β-片层结构可富集Aβ蛋白使其聚集成不溶性纤维,使其不易被蛋白酶降解,从而形成沉淀,继而生成老年斑。最终使得神经细胞内的Ca2+浓度增加,激活蛋白激酶,导致Tau蛋白的异常磷酸化。转基因动物模型研究表明:其实在不溶性斑块与可溶性Aβ蛋白之间,不论在血脑屏障内、外都存在一种受多种因素调节的双向动态平衡机制,而AD的发生可能正是因为这种平衡被打破。而不溶性斑块即NFTs的形成过程中Ca2+通过调节钙结合蛋白和其激酶来发挥作用。研究表明,微管的变化会影响细胞中的Ca2+的稳定性,所以这是一个双向的作用。Aβ蛋白产生细胞损伤甚至死亡的具体机制为:①产生氧化损伤,②激发免疫反应。由不溶性Aβ蛋白聚集而成的纤维正是导致AD的一大原因。它的沉积可以引起多种炎症反应,从而导致广泛的神经元变性甚至死亡。而当海马体的神经元变性或者死亡时,就会导致记忆和认知的障碍。
2.3 Tau蛋白与ApoE的关系 APOE是一种载体蛋白,参与脂蛋白的转化与代谢过程,其基因可以调节许多生物学功能。其实,不但异常过度磷酸化的Tau蛋白会富集成NFTs。ApoE与Tau蛋白之间的相互作用也会形成NFTs。ApoE有三种异构体,分别是ApoE2,ApoE3和ApoE4,分别由19号染色体上的ε2,ε3,ε4三个等位基因所编码。有体外试验证明,ApoE3可与Tau蛋白按1:1的生物比例形成稳定的十二烷基硫酸钠(SDS)类化合物,而ApoE4与Tau蛋白无明显作用[7],所以ApoE3比ApoE4与Tau蛋白有更好的亲和力。在实验中,从脑内提取的磷酸化Tau蛋白会抑制ApoE3与其之间的相互作用。因为APOE3与Tau蛋白的良好亲和力,其与Tau蛋白的结合会防止Tau蛋白被过度异常磷酸化。而ApoE4由于它不可以与Tau蛋白之间相互作用,所以这就导致Tau蛋白无法被保护,从而被过度异常磷酸化形成PHFs继而产生NFTs,最终导致神经元骨架被破坏,使神经元变性甚至死亡。此外有研究表明ApoE其实是通过与其受体结合从而去抑制与Tau蛋白相关的激酶的,如P35,P-GSK-3β,CDK5等,继而调控着神经元内Tau蛋白的磷酸化水平。有研究人员认为在斑块区域附近的ApoE可以通过它的受体传递一种慢性信号,这种信号会使激酶的某些磷酸化底物的含量上升或者下降,从而最终使得Tau蛋白磷酸化程度的改变。APOE不同的异构体所表达的信号强弱与功能也不一样,比如ApoE2可以降低Tau蛋白的磷酸化;而ApoE4却是可以促进Tau蛋白的磷酸化[8]。
3. Tau蛋白与AD的关系
异常过度磷酸化的Tau蛋白的堆积以及沉降,从而引起神经元变性死亡的积累是导致AD的一个重要原因。而Tau蛋白的异常过度磷酸化是由蛋白激酶活性增高或磷酸酯酶活性降低所导致的。目前,AD患者的脑中已分离出三种Tau蛋白,他们分别是胞浆正常Tau蛋白(C-Tau)、可溶于水的异常磷酸化Tau蛋白(AD p-Tau)和异常修饰聚集为PHF的Tau蛋白(PHF-Tau)[9]。其中,PHF是NFTs的主要成分,PHF-Tau彼此相连形成混合微丝,表现出不溶解性和对蛋白酶解的抵抗性。
在实验模型中,异常磷酸化的蛋白的聚集对神经元具有毒性。此外,已有学者提出致病型Tau蛋白在神经元之间的传播是导致AD在脑中扩散的原因;随着AD进展,这种扩散会遵循一种独特的进展方式穿过脑部区域[10~12]。
也有学者认为,Tau沉积梯度的变化可能导致了AD的发生。因为在正常的轴突里,Tau的沉积在接近轴突末端增加,但在变性的轴突中Tau的沉积梯度是相反的,即在轴丘Tau的沉积最多。
4.检测与治疗
在检测方面,疑似患者的脑脊液(CSF)中磷酸化Tau蛋白浓度的增加和使用核磁共振技术显示的脑萎缩加重情况在发病前约15年就会出现,认知功能障碍仅在临床诊断前约5年时才检出,所以我们不能只靠认知功能障碍的检测去判断。往往出现认知功能障碍时神经元已受到不可逆的损伤。
而近年来,Tau PET示踪剂这一追踪技术的发现大大提高了AD的可鉴别性。Tau PET示踪剂是非毒性、亲脂性的小分子,能快速穿过血脑屏障,并迅速从血中清除,具有选择性、特异性和可逆性靶向结合的特征。PET示踪剂的选择必须由靶目标上有效的结合位点浓度决定,Tau示踪剂根据其与Tau蛋白的亲合力可分为选择性与非选择性,选择性TauPET示踪剂应该只能与一种Tau超微结构结合,并具有特异性和高结合性,对AD而言,高选择性的TauPET示踪剂必须能竞争性阻断与Tau配体相结合的Aβ浓聚物。示踪剂的这种特异病征性和局限性分布,有助于对AD及各种与Tau相关疾病的鉴别[13]。
在治疗方面,近年来发现了一种质子泵抑制剂,至少有2项研究表明已发现了使用质子泵抑制剂(proton pump inhibitor,PPI)和新发痴呆之间存在有统计学意义的关联[14,15],但仍需要进一步研究以确定该关联是否存在因果关系。一项纳入73,000多例75岁及75岁以上基线时无痴呆成人的前瞻性队列研究表明,定期使用PPI与新发痴呆的风险增加相关(HR 1.44,95%CI 1.36~1.52),并且在校正了多种潜在的混杂因素(包括年龄、性别、抑郁、脑卒中、心脏病及多药治疗)后,这种关联仍然具有统计学意义[15]。一项较小型研究也发现全因痴呆(HR 1.3)和AD痴呆(HR 1.4)与PPI使用均存在相似的关联[14]。
有限的介入治疗前的数据表明PPI与淀粉样物质和Tau蛋白之间均存在相互作用关系,这些数据提供了可能的生物学合理性[16~18]。长期使用PPI所引起的维生素B12或其他营养素的吸收不良也可能与其有一定关系[19]。另一方面,这种关联性可能反应了一些因素所引起的残余混杂影响,即一些与PPI同时使用和痴呆的发生均相关的因素,还需要更多的研究来确认[20]。
1 刘倩,吴为辉,李人望,等. 载脂蛋白E与阿尔兹海默病的相关研究进展[J]. 化学进展,2007,19(12): 2006-2011.
2 De Strooper B. Proteases and proteolysis in Alzheimer disease: a multifactorial view on the disease process[J]. Physiol Rev,2010,90(2): 465-494. DOI:10.1152/physrev.00023.2009.
3 Hoe HS,Freeman J,Rebeck GW. Apolipoprotein E decreases tau kinases and phospho-tau levels in primary neurons[J]. Mol Neurodegener,2006,1: 18. DOI:10.1186/1750-1326-1-18.
4 崔行. Tau蛋白与老年性痴呆[J]. 山东医药,2000,40(20): 51-52. DOI:10.3969/j.issn.1002-266X.2000.20.039.
5 Majounie E,Cross W,Newsway V,et al. Variation in tau isoform expression in different brain regions and disease states[J]. Neurobiol Aging,2013,34(7): 1922-1927. DOI:10.1016/j.neurobiolaging.2013.01.017.
6 Drubin DG,Kirschner MW. Tau protein function in living cells[J]. J Cell Biol,1986,103(6 Pt 2): 2739-2746. DOI:10.1083/jcb.103.6.2739.
7 Klarenbeek J,Goedhart J,van Batenburg A,et al. Fourth-generation epac-based FRET sensors for cAMP feature exceptional brightness,photostability and dynamic range: characterization of dedicated sensors for FLIM,for ratiometry and with high affinity[J]. PLoS ONE,2015,10(4): e0122513. DOI:10.1371/journal.pone.0122513.
8 Andrews-Zwilling Y,Bien-Ly N,Xu Q,et al. Apolipoprotein E4 causes age- and Tau-dependent impairment of GABAergic interneurons,leading to learning and memory deficits in mice[J]. J Neurosci,2010,30(41): 13707-13717. DOI:10.1523/JNEUROSCI.4040-10.2010.
9 Mondragón-Rodríguez S,Basurto-Islas G,Lee HG,et al. Causes versus effects: the increasing complexities of Alzheimer’s disease pathogenesis[J]. Expert Rev Neurother,2010,10(5): 683-691. DOI:10.1586/ern.10.27.
10 Guo JL,Lee VM. Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer-like tangles[J]. J Biol Chem,2011,286(17): 15317-15331. DOI:10.1074/jbc.M110.209296.
11 Iba M,Guo JL,McBride JD,et al. Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer’s-like tauopathy[J]. J Neurosci,2013,33(3): 1024-1037. DOI:10.1523/JNEUROSCI.2642-12.2013.
12 Medina M,Avila J. The role of extracellular Tau in the spreading of neurofibrillary pathology[J]. Frontiers in Cellular Neuroscience,2014,8. DOI:10.3389/fncel.2014.00113.
13 何跃,刘丹. tau 蛋白在阿尔茨海默病中的病理生理机制及 tau 示踪剂研究现状[J]. 神经疾病与精神卫生,2016,16(2): 239-242. DOI:10.3969/j.issn.1009-6574.2016.02.034.
14 Haenisch B,von Holt K,Wiese B,et al. Risk of dementia in elderly patients with the use of proton pump inhibitors[J]. Eur Arch Psychiatry Clin Neurosci,2015,265(5):419-428.DOI:10.1007/s00406-014-0554-0.15 Gomm W,von Holt K,Thomé F,et al. Association of Proton Pump Inhibitors With Risk of Dementia: A Pharmacoepidemiological Claims Data Analysis[J]. JAMA Neurol,2016,73(4): 410-416. DOI:10.1001/jamaneurol.2015.4791.
16 Rojo LE,Jans A-M,Saavedra IN,et al. Selective interaction of lansoprazole and astemizole with tau polymers: potential new clinical use in diagnosis of Alzheimer’s disease.[J]. Journal of Alzheimer’s Disease,2010(2): 573-589. DOI:10.3233/JAD-2010-1262.
17 Badiola N,Alcalde V,Pujol A,et al. The proton-pump inhibitor lansoprazole enhances amyloid beta production[J]. PLoS ONE,2013,8(3): e58837. DOI:10.1371/journal.pone.0058837.
18 Fallahzadeh MK,Borhani Haghighi A,Namazi MR. Proton pump inhibitors: predisposers to Alzheimer disease[J]. J Clin Pharm Ther,2010,35(2): 125-126. DOI:10.1111/j.1365-2710.2009.01100.x.
19 Lam JR,Schneider JL,Zhao W,et al. Proton pump inhibitor and histamine 2 receptor antagonist use and vitamin B12 deficiency[J]. JAMA,2013,310(22): 2435-2442. DOI:10.1001/jama.2013.280490.
20 Kuller LH. Do Proton Pump Inhibitors Increase the Risk of Dementia[J]. JAMA Neurol,2016,73(4): 379-381. DOI:10.1001/jamaneurol.2015.4931.
Related research on the phosphorylation of Tau protein effect on Alzheimer’s disease
(LIU Yanyi, CHEN Chao, LI Zhipeng, YANG Ze.
Institute of Geriatrics, Chinese Ministry of Health, Beijing Hospital, Beijing 100730, China.)
Alzheimer’s disease is a kind of common chronic progressive neurodegenerativing disease,primarily manifested as irreversible whole brain damage.Because of complicated factors,nosogenesis has not been determined.One kind of theory is that abnoraml Tau protein phosphorylation and straight filament will lead to NFTs.This caused microtubule deformation in the neurofibrillary .Then neuronal will degeneration and even death.When lots of neuronal death that would trigger AD.Therefore it’s important to find out the mechanism of abnormal phosphorylation of Tau protein and then targeted therapy for patients.
Alzheimer’s disease, Tau protein, NFTs
1.北京医院卫生部老年医学研究所,卫生部老年医学重点实验室 100730
2.北京师范大学珠海分校 519087
国家自然科学基金(81061120527,81370445,81472408),卫生部公益性研究基金(201302008)和国家科技部十二五支撑计划(2012BAI10B01,2015BAI06B03)。
10.3969/j.issn.1672-4860.2016.05.002
2016-9-17
猜你喜欢
中国骨质疏松杂志(2022年9期)2022-10-18
石油地质与工程(2022年4期)2022-08-06
——水芹主要害虫识别与为害症状
长江蔬菜(2022年13期)2022-07-29
石油化工应用(2022年3期)2022-04-20
化工管理(2021年23期)2021-08-25
土壤与作物(2021年2期)2021-06-01
西安石油大学学报(自然科学版)(2021年1期)2021-01-27
科学(2020年2期)2020-08-24
中国癌症防治杂志(2019年5期)2019-01-04
中国循环杂志(2015年10期)2015-12-24
