
病毒性心肌炎小鼠心肌中MMP-2和NF-κB的表达*
2016-10-26 05:35阮妙华周爱华褚茂平
中国病理生理杂志 2016年9期
阮妙华, 王 凯, 王 丹, 周爱华, 褚茂平, 陈 其△, 钱 燕
(温州医科大学 1附属第一医院儿童内科, 2附属第二医院儿童心血管科,浙江 温州 325027)
病毒性心肌炎小鼠心肌中MMP-2和NF-κB的表达*
阮妙华1, 王 凯1, 王 丹1, 周爱华1, 褚茂平2, 陈 其2△, 钱 燕1
(温州医科大学1附属第一医院儿童内科,2附属第二医院儿童心血管科,浙江 温州 325027)
目的: 初步探讨基质金属蛋白酶2(matrix metalloproteinase-2,MMP-2)和核因子κB(nuclear factor-κB,NF-κB)p65在柯萨奇病毒B3(CVB3)诱导的小鼠病毒性心肌炎心肌中的表达。方法: 6周龄近交系雄性 BALB/c小鼠随机分为对照组和病毒性心肌炎组,分别给予0.1 mL PBS和10-5.69TCID50/mL CVB3注射,第 4天和第10天各随机取 8只小鼠处死并取血和心脏标本。Reed-Muench法测定接种病毒滴度;苏木素伊红(hematoxylin and eosin,HE)染色后光镜观察心肌组织病理学改变;应用免疫组化法(immunohistochemistry,IHC)检测MMP-2和NF-κB p65在心肌组织表达的分布和含量;用Western blot法检测MMP-2、NF-κB p65和IκBα在心肌组织的表达,用ELISA检测血清TNF-α含量的变化。结果: 与对照组相比,免疫组化和Western blot实验结果提示病毒性心肌炎小鼠心肌中MMP-2和NF-κB p65的表达显著升高(P<0.05);感染组IκBα的表达呈下降趋势(P<0.05);ELISA结果提示血清TNF-α含量在感染组显著升高,差异有统计学显著性(P<0.05)。结论: 病毒性心肌炎小鼠心肌组织中TNF-α、NF-κB p65和MMP-2表达显著上调,可能通过相关机制参与疾病的发生。
心肌炎; 基质金属蛋白酶2; 核因子κB
人感染柯萨奇病毒B3(coxsackievirus B3,CVB3)可导致病毒性心肌炎(viral myocarditis,VM),引起心肌组织的实质性损伤,严重者可导致心力衰竭甚至死亡,其中约10%~20%的病人可进展至扩张性心肌病[1]。目前关于病毒性心肌炎的发病机制尚不明确,考虑主要由病毒直接侵害和炎症反应所致。研究报道[2],炎症因子肿瘤坏死因子α(tumor necrosis factor-α,TNF-α)可通过诱导基质金属蛋白酶(matrix metalloproteinases,MMPs)的表达及活性上调,降解细胞外基质(extracellular matrix,ECM),介导病毒性心肌炎的病理生理过程。MMP-2属于MMPs家族的明胶酶类,作用底物最广泛。据报道,其上游信号分子核因子κB(nuclear factor-κB,NF-κB) p65的核易位引起活化后,结合MMPs的启动子序列,从而使MMPs活性和表达上调[3]。目前,国内外TNF-α/NF-κB/MMPs通路的上下级联关系在A549等细胞模型中得到证实[3-5],但在CVB3诱导的病毒性心肌炎中未见相关报道。本实验旨在通过CVB3诱导的心肌炎模型中,探讨病毒性心肌炎中TNF-α对MMP-2表达的影响及其作用途径,从而为进一步阐明病毒性心肌炎的发病机制提供科学依据,并为临床深入认识和治疗VM提供新思路。
材 料 和 方 法
1 材料
CVB3 Nancy标准株购自山东省医学科学院病毒室。近交系雄性BALB/c小鼠,4~6周龄,购自上海斯莱克实验动物有限责任公司。随机分为对照组(n=25)和 病毒性心肌炎组(n=30)。HE染色试剂盒、PBS粉剂、柠檬酸修复液、苏木素染液、中型树胶及浓缩型DAB试剂盒、兔源MMP-2多克隆抗体、小鼠的TNF-α ELISA试剂盒、即用型SABC过氧化物酶、抗MMP-2和NF-κB p65、IκBα抗体由北京生物合成有限公司提供;乙醇及二甲苯等试剂为市售产品。
2 方法
2.1 动物模型的建立 动物的感染按照Hua等[6]报道的方法进行,BALB/c的小鼠经腹腔接种含10-5.69TCID50/mL的CVB3液0.1 mL,对照组小组接种相同剂量的PBS。
2.2 标本的采集 分别于病毒感染后的第 4天、10天各随机取 8只小鼠处死并取血测TNF-α的浓度;取心脏标本做病理组织学检测和Western blot实验。
2.3 组织病理学检查 常规处理心脏标本,HE染色,光镜下观察心肌的病理变化。
2.4 血清TNF-α水平的检测 应用TNF-α ELISA试剂盒按说明书进行。
2.5 免疫组化SABC法和Western blot法检测心肌组织MMP-2、IκBα以及NF-κB p65蛋白的表达 心肌组织石蜡切片经烤片,脱水,抗原修复,滴加MMP-2 的 I 抗(1∶50)、IκBα和NF-κB p65的 I 抗(1∶100),4 ℃过夜,依次加入相对应的 II 抗,试剂SABC,DAB显微镜下显色,苏木素复染,脱水,中性树胶封片,镜检。蛋白阳性表达在镜下为棕黄色,用Image-Pro Plus 6.0图像分析软件测定棕黄色颗粒平均吸光度来代表相关蛋白表达量。同时心肌切片常规苏木精-伊红染色依照细胞蛋白抽提试剂盒说明书提取细胞蛋白。采用BCA蛋白质的测定蛋白检测试剂盒(碧云天生物技术研究所)。蛋白质样品于99 ℃煮沸10 min,进行SDS-PAGE分离后转膜,随后,膜在室温下浸入5%脱脂牛奶30 min后,I 抗MMP-2(1∶1 000稀释)、IκBα(1∶500稀释)和 NF-κB p65(1∶500稀释)抗体4 ℃过夜。TBST清洗NC膜,每次10 min,共3次。然后,NC膜浸泡于相对应的 II 抗(1∶2 000),室温孵育1~2 h,再次清洗后显色。用ImageJ图像分析软件对Western blot条带进行定量分析,计算MMP-2、IκBα和NF-κB 与β-actin吸光度的比值,得到其相对表达量。
3 统计学处理
数据均采用均数±标准差(mean±SD表示),采用 SPSS 16.0统计软件,所有数据均进行正态性检验和方差齐性检验,在方差齐的基础上行单因素方差分析,并用Bonferroni校正的t检验对各组均数进行两两比较。以P<0.05为差异有统计学意义。
结 果
1 心肌炎模型的评价
小鼠在接种病毒后出现体重下降、活动减少,第5天开始出现死亡,第10天达死亡高峰,总共死亡9只;而正常对照组体重增加、活动良好,无死亡。接种病毒第10天心肌细胞开始出现混浊肿胀和溶解坏死,炎性细胞呈灶状浸润,心肌肿胀坏死,而对照组未见这些改变,因此,本实验成功建立小鼠CVB3病毒性心肌炎模型,见图1。
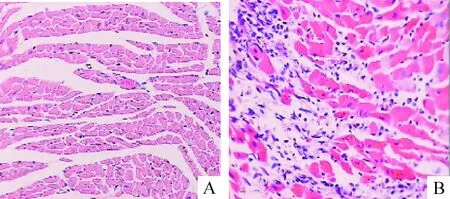
Figure 1.Observation of the myocardium with hematoxylin and eosin staining (×200). A: normal tissues in control group, no inflammation was observed; B: CVB3 infection for 10 d, the myocardium with inflammation and cell necrosis.
图1 感染组和对照组心肌组织HE染色的比较
2 各组血清TNF-α含量的比较
与正常组相比,心肌炎感染第4天血清TNF-α的水平明显升高,为(25.9±0.5)ng/L。感染第10天血清TNF-α的含量显著升高,为(31.0±0.7)ng/L,正常对照组的血清TNF-α含量为(16.1±0.1)ng/L。感染组血清TNF-α与阴性对照组相比,含量明显提高,差异有统计学显著性(P<0.05),见图2。
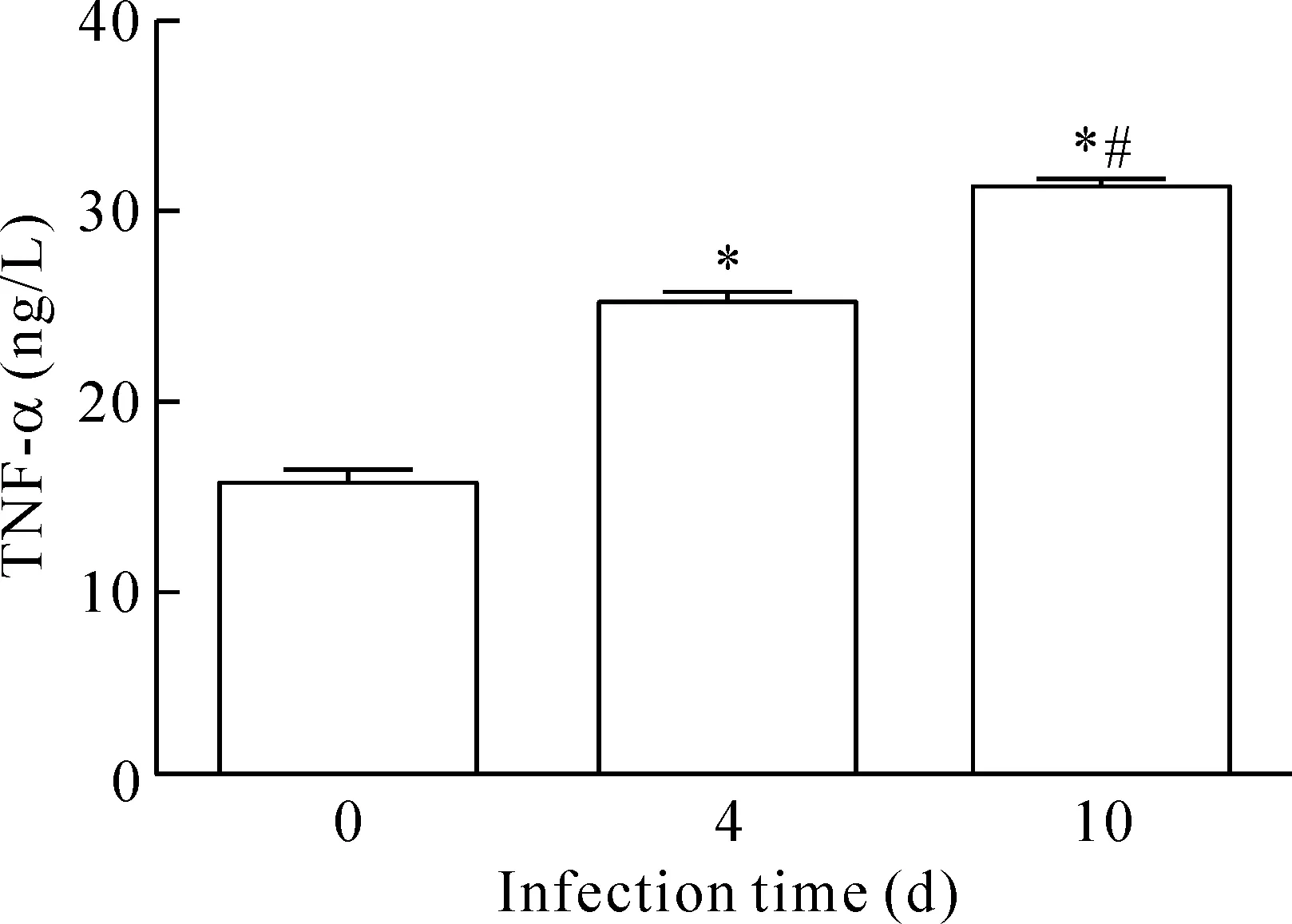
Figure 2.The concentration of TNF-α was higher after development of myocarditis by infection with CVB3. Mean±SD.n=8.*P<0.05vs0 d;#P<0.05vs4 d.
图2 对照组和感染组血清TNF-α含量的比较
3 心肌MMP-2、NF-κB p65和IκBα蛋白表达的变化
在正常对照组,MMP-2和NF-κB p65为弱表达,而第4天开始表达增加,第10天达高峰。IκBα在正常对照组中可见表达,第4天开始出现表达下降,第10天下降最明显。MMP-2和NF-κB 在感染组与对照组相比表达显著升高,差异有统计学显著性;而IκBα的表达在感染组显著下降,与对照组相比,差异有统计学显著性(P<0.05),见图3、4。
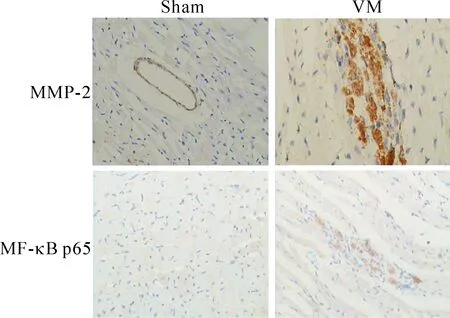
Figure 3.The images of immunohistochemistry for observing the expression of MMP-2 and NF-κB p65 (×20).
图3 心肌组织MMP-2及NF-κB p65的变化
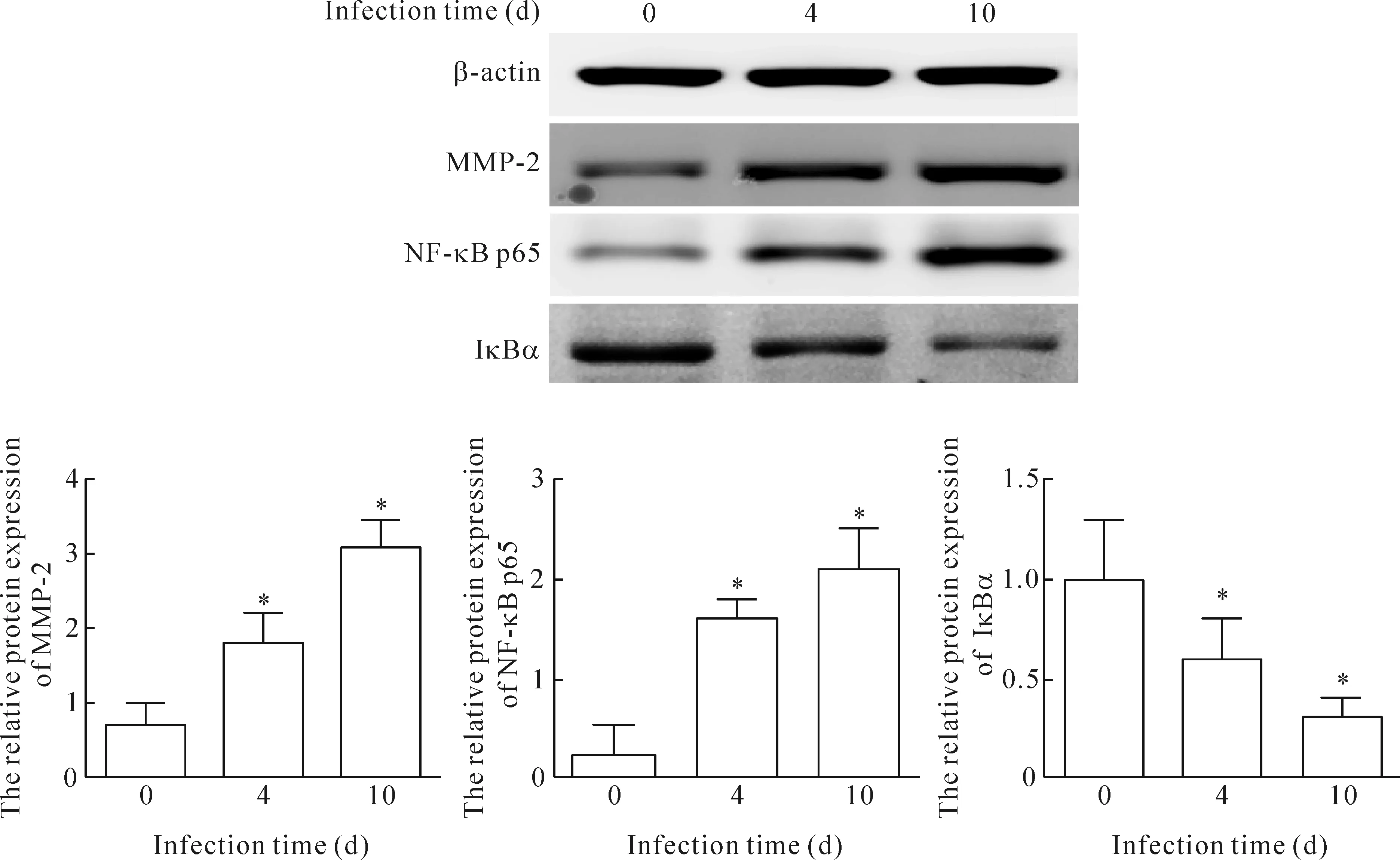
Figure 4.The effects of CVB3 infection on the protein expression of MMP-2, NF-κB p65 and IκBα in the myocardium. Mean±SD.n=8.*P<0.05vs0 d.
图4 MMP-2、NF-κB p65和IκBα在心肌中的表达
讨 论
本实验在CVB3诱导的小鼠病毒性心肌炎模型中发现,在感染组第4天开始,心肌组织MMP-2、NF-κB p65和TNF-α开始上调,感染后第10天上调更显著。Malz等[7]报道,在CVB3诱导的病毒性心肌炎模型中,MMPs的mRNA表达以及酶普法检测到活性显著提高,可降解细胞外基质成分,介导炎症反应以及心肌重塑,从而参与心肌炎的病理生理过程,与我们实验结果相一致。
病毒性心肌炎的发病机制目前仍未明确,目前认为主要是由于病毒直接损害和免疫变态反应所致。近年来的研究表明[8-10],VM的心肌病变(炎症细胞浸润和坏死)与炎症细胞因子TNF-α的水平呈正相关。VM中TNF-α增加的原因目前仍不清楚,推测可能是多种原因所致[11-14]:(1)与病毒感染直接相关;(2)VM心肌的单核细胞、淋巴细胞等炎性细胞可合成TNF-α,另外可能与巨噬细胞被活化有关;(3)小鼠接种心肌炎病毒后,NK细胞活性升高,而NK细胞也能合成TNF-α。在我们的研究中发现, TNF-α的血清浓度在感染后第4天开始显著升高,在第10天达到高峰,因此,TNF-α可能参与病毒性心肌炎的病理生理过程,但具体作用机制目前仍不明确。目前国内外的研究提及 TNF-α主要通过参与平滑肌细胞迁移、炎性细胞浸润以及MMPs表达,从而参与心肌炎的病理生理机制过程。
MMPs是一类依赖锌离子和钙离子的内源性蛋白水解酶家族,能特异性降解细胞外基质ECM成分,从而参与病毒性心肌炎的病理生理过程。明胶酶包括MMP-2和MMP-9属于MMPs的一大类,MMP-2和MMP-9两者作用相似,因为作用底物广泛。MMP-2可由血管内皮细胞、心肌坏死灶以及周围的心肌细胞等分泌。在我们的实验中,CVB3感染第4 天时可以看到MMP-2的表达显著增加,第10天达到高峰。我们推测在CVB3病毒性心肌炎模型中MMP-2表达增加,通过参与心肌炎ECM的降解,介导炎症反应。
TNF-α主要是通过作用于MMPs的启动子序列,增强MMPs的表达及活性上调。而MMPs可以通过参与TNF-α的细胞膜受体水解,从而活化TNF-α,因此两者之间存在相互作用机制。在CVB3诱导的心肌炎症反应中如TNF-α等炎性因子,主要是通过激活NF-κB p65核易位起作用。NF-κB p65主要是异二聚体的方式存在于细胞质中,通过与IκBα结合,当IκBα降解后,p65异二聚体从细胞质进入细胞核中,与MMPs的启动子序列结合,上调相关表达。我们的研究也证实了,感染组NF-κB p65的核表达显著增加,相应的,细胞质中IκBα因为降解而表达下调。在其它细胞模型中证实[11-15],TNF-α通过NF-κB上调MMPs的表达,并且给予TNF-α的抑制剂可下调MMPs的表达,因此,明确了TNF-α和MMPs之间的上下级联关系。但是,TNF-α诱导MMPs活化的过程中是否有别的通路共同参与介导心肌炎的病理生理过程呢?Haudek等[16]报道,给予TNF-α急性刺激,心肌NF-κB(p50-p65)二聚体可快速激活转运入心肌细胞核,而给予慢性TNF-α刺激不仅使NF-κB(p50-p65)异二聚体转录激活并且伴有NF-κB(p50-p65)同二聚体的失活。
综上所述,我们在病毒性心肌炎模型中发现TNF-α、NF-κB和MMP-2存在表达上调,并且 NF-κB 的相关激酶IκBα在细胞质中的表达呈相关性下调,由此推测TNF-α/NF-κB/MMP-2通路的活化可能是其机制之一。然而,NF-κB不同异构体在TNF-α/NF-κB/MMPs该路径中到底起什么样的作用,同时不同浓度的TNF-α对该路径又会产生什么样的影响,仍需进一步深入研究。
[1] Pollack A, Kontorovich AR, Fuster V, et al. Viral myocarditis: diagnosis, treatment options, and current controversies[J]. Nat Rev Cardiol, 2015, 12(11):670-680.
[2] Wang Y, Li DL,Zhang XB, et al. Increase of TNF-α-stimulated osteoarthritic chondrocytes apoptosis and decrease of matrix metalloproteinases 9 by NF-κB inhibition[J]. Biomed Environ Sci, 2013, 26(4):277-283.
[3] Lin CC, Tseng HW, Hsieh HL, et al. Tumor necrosis factor-α induces MMP-9 expression via p42/p44 MAPK, JNK, and nuclear factor-κB in A549 cells[J]. Toxicol Appl Pharmacol, 2008, 229(3):386-398.
[4] Chtourou Y, Fetoui H, Jemai R, et al. Naringenin reduces cholesterol-induced hepatic inflammation in rats by modulating matrix metalloproteinases-2, 9 via inhibition of nuclear factor κB pathway[J]. Eur J Pharmacol, 2014, 746:96-105.
[5] Zhu X, Wang Z, Hu C, et al. Honokiol suppresses TNF-α-induced migration and matrix metalloproteinase expression by blocking NF-κB activation via the ERK signaling pathway in rat aortic smooth muscle cells[J]. Acta Histochem, 2014, 116(4):588-595.
[6] Hua W, Chen Q, Gong F, et al. Cardioprotection of H2S by downregulating iNOS and upregulating HO-1 expression in mice with CVB3-induced myocarditis[J]. Life Sci, 2013, 93(24):949-954.
[7] Malz R, Weithauser A, Tschöpe C, et al. Inhibition of coagulation factor Xa improves myocardial function during CVB3-induced myocarditis[J]. Cardiovasc Ther, 2014, 32(3):113-119.
[8] Ahn J, Kim J. Mechanisms and consequences of inflammatory signaling in the myocardium[J]. Curr Hypertens Rep, 2012, 14(6):510-516.[9] Wang D,Chen Y, Jiang J, et al. Carvedilol has stronger anti-inflammation and anti-virus effects than metoprolol in murine model with coxsackievirus B3-induced viral myocarditis[J]. Gene, 2014, 547(2):195-201.
[10]Sun XH, Fu J, Sun DQ. Halofuginone alleviates acute viral myocarditis in suckling BALB/c mice by inhibiting TGF-β1[J]. Biochem Biophys Res Commun, 2016, 473(2):558-564.
[11]Gullestad L, Ueland T, Vinge LE, et al. Inflammatory cytokines in heart failure: mediators and markers[J]. Car-diology, 2012, 122(1):23-35.
[12]Jeong JW, Kim JW, Ku SK, et al. Essential oils purified from Schisandrae semen inhibits tumor necrosis factor-α-induced matrix metalloproteinase-9 activation and migration of human aortic smooth muscle cells[J]. BMC Complement Altern Med, 2015, 15:7.
[13]王桂君, 姚玉胜, 王洪新. 肿瘤坏死因子α通过PI3K-IP3R-Ca2+途径诱导乳鼠心肌肥大[J]. 中国病理生理杂志, 2016, 32(1):21-26.
[14]张淑芬, 杨红军, 邓兵梅,等. TNF-α在 CCD模型大鼠背根神经节中的表达及机制[J]. 中国病理生理杂志, 2015, 31(4):675-679.
[15]Cao Q, Jiang Y, Shi J, et al. Artemisinin inhibits the proliferation, migration, and inflammatory reaction induced by tumor necrosis factor-α in vascular smooth muscle cells through nuclear factor kappa B pathway[J]. J Surg Res, 2015, 194(2):667-678.
[16]Haudek SB, Bryant DD, Giroir BP. Differential regulation of myocardial NF kappa B following acute or chronic TNF-alpha exposure.[J]. J Mol Cell Cardiol, 2001, 33(6):1263-1271.
(责任编辑: 卢 萍, 罗 森)
Expression of MMP-2 and NF-κB in myocardium of mice with viral myocarditis
RUAN Miao-hua1, WANG Kai1, WANG Dan1, ZHOU Ai-hua1, CHU Mao-ping2, CHEN Qi2, QIAN Yan1
(1DepartmentofPediatrics,TheFirstAffiliatedHospital,2DepartmentofPediatricCardiology,TheSecondAffiliatedHospital,WenzhouMedicalUniversity,Wenzhou325027,China.E-mail:cqq57@126.com)
AIM: To observe the effects of TNF-α/nuclear factor-κB(NF-κB)/matrix metalloproteinase-2(MMP-2) pathway on the expression of MMP-2 in the mice with viral myocarditis. METHODS: Six-week-old inbred male mice were randomly assigned to control and myocarditis group. The mice in myocarditis group and control group were intraperitoneally inoculated with 0.1 mL 10-5.69TCID50/mL coxsackievirus B3 and vehicle (PBS), respectively. Ten mice were sacrificed at the 4th and 10th days after injection. The blood and heart specimens were harvested. The serum content of TNF-α was measured by ELISA. The myocardial levels of MMP-2, NF-κB p65 and IκBα were determined by Western blot. RESULTS: Compared with control group, the protein expression of MMP-2 and NF-κB p65 in the myocardium and the serum content of TNF-α were significantly increased in myocarditis group (P<0.05). The protein expression of IκBα was lower in myocarditis group than that in control group (P<0.05). CONCLUSION: TNF-α, NF-κB p65 and MMP-2 were higher in the mice with acute viral myocarditis. The increased expression of them might be involved in the pathogenesis of viral myocarditis.
Myocarditis; Matrix metalloproteinase-2; Nuclear factor-κB
杂志网址: http://www.cjpp.net
1000- 4718(2016)09- 1704- 05
2016- 04- 28
2016- 07- 09
温州市科技局基金项目(No. Y20150142)
△通讯作者 Tel: 13957766274; E-mail: cqq57@126.com
R392.3; R363
A
10.3969/j.issn.1000- 4718.2016.09.029
猜你喜欢
湖南畜牧兽医(2021年6期)2022-01-24
湖南饲料(2021年4期)2021-10-13
食品安全导刊(2021年21期)2021-08-30
文萃报·周二版(2021年11期)2021-04-06
天津医科大学学报(2019年6期)2019-08-13
中国人兽共患病学报(2018年7期)2018-07-31
中国医药指南(2017年3期)2017-11-13
中国继续医学教育(2015年4期)2016-01-07
中国体外循环杂志(2015年3期)2015-12-08
中国民族民间医药·下半月(2014年2期)2014-09-26
