
阿托伐他汀钙片有关物质的HPLC分析法
2016-09-22 07:24于妮娜孟淑华
西北药学杂志 2016年5期
于妮娜,张 玲,孟淑华
(1.陕西省新药审评中心,西安 710065;2.西安天一秦昆制药有限责任公司,西安 710077)
阿托伐他汀钙片有关物质的HPLC分析法
于妮娜1,张玲2,孟淑华2
(1.陕西省新药审评中心,西安710065;2.西安天一秦昆制药有限责任公司,西安710077)
目的 建立阿托伐他汀钙片有关物质的测定方法。方法采用反相HPLC法,测定阿托伐他汀钙片有关物质的含量。采用SyncronisC18色谱柱(250mm×4.6mm,5μm);流动相A为醋酸缓冲液∶乙腈∶四氢呋喃(61∶27∶12),流动相B为醋酸缓冲液∶乙腈∶四氢呋喃(47∶41∶12);流速为1.5mL·min-1;检测波长为260nm;柱温为35 ℃。结果阿托伐他汀钙杂质1、杂质2、杂质3、杂质4、杂质6与阿托伐他汀峰均可达到基线分离;检测限分别为阿托伐他汀2ng,杂质1 2ng、杂质2 2ng、杂质3 2ng、杂质4 2ng、杂质6 1.6ng,且重复性良好,总杂质RSD值为1.87%(n=6)。结论该方法稳定、可靠、专属性强,可用于阿托伐他汀钙片的有关物质检测。
阿托伐他汀钙;有关物质;杂质测定;高效液相色谱法
阿托伐他汀钙(C33H34FN2O5)2Ca·3H2O是选择性、竞争性3-羟基-3-甲基戊二酰辅酶A(HMG-CoA)还原酶抑制剂,可显著降低胆固醇水平,降低心肌梗死和脑卒中的发病危险,对原发性高胆固醇血症,包括家族性高胆固醇血症、混合型高胆固醇血症以及纯合子家族性高胆固醇血症患者具有显著的疗效。对于阿托伐他汀钙片有关物质的研究也有文献报道[1-4],而国家标准[5-6]仅控制单个杂质和总杂质,对于杂质的研究并不充分,不能有效地控制。本实验结合USP、CP和EP等[7-8]对阿托伐他汀钙片有关物质的检测方法进行研究,采用对照品外标法对已知杂质进行定性、定量测定,其他未知杂质采用主成分自身对照法定量。本实验所建立方法可使阿托伐他汀钙、5个已知杂质及未知杂质达到基线分离,且方法简便易行、结果准确。
1 仪器与试药
1.1仪器U3000型高效液相色谱仪(美国戴安ThermoSeparationProducts公司);BS110S型分析天平,CP225D型分析天平(德国赛多利斯公司);色谱柱:SyncronisC18(250mm×4.6mm,Thermo公司);C18柱(250mm×4.6mm,粒径5μm,江苏汉邦公司);XB-C18柱(250mm×4.6mm,粒径5μm,Welchmaterials公司)。
1.2试药色谱纯:乙腈,四氢呋喃(TEDIA公司);冰醋酸(江苏汉邦公司)。分析纯:柠檬酸,氨水(天津市登峰化学试剂厂);醋酸铵(郑州派尼化学试剂厂)。水为超纯水。
2 方法与结果
2.1色谱条件用十八烷基硅烷键合硅胶为填充剂。流动相A:醋酸缓冲液(取醋酸铵3.9g,加水1 000mL,用冰醋酸调节pH值为5.0~5.1)∶乙腈∶四氢呋喃(61∶27∶12);流动相B:醋酸缓冲液∶乙腈∶四氢呋喃(47∶41∶12);按照下列程序梯度洗脱[9],见表1,进样前平衡10min。流速:1.5mL·min-1;检测波长:260nm;柱温:35 ℃。
2.2系统适用性实验取阿托伐他汀钙及阿托伐他汀杂质2对照品适量,加混合溶剂[以0.05mol·L-1的柠檬酸铵溶液(0.05mol·L-1柠檬酸用氨水调节pH值至7.4)-乙腈(50∶50)]制成质量浓度为50μg·mL-1的溶液,取20μL注入液相色谱仪,理论板数以阿托伐他汀计应不低于5 000,分离度应符合要求。见图1。
表1梯度洗脱顺序
Tab.1Thegradientelutionprogram
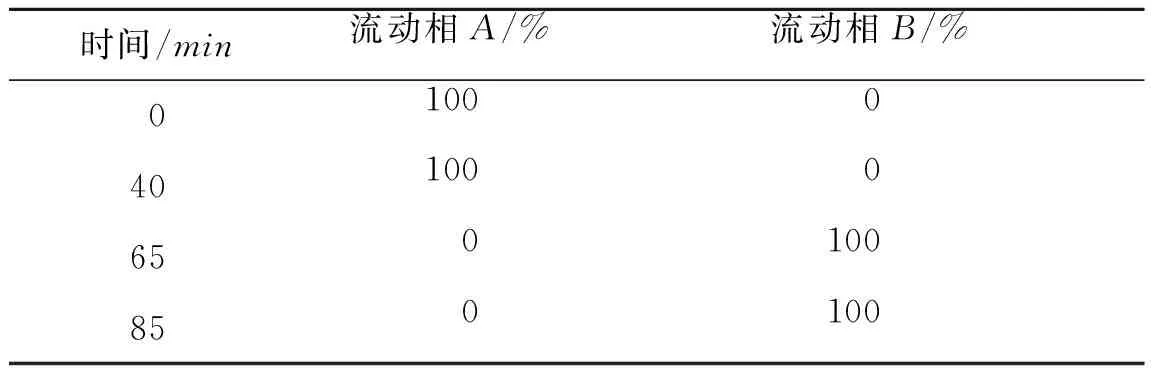
时间/min流动相A/%流动相B/%01000401000650100850100
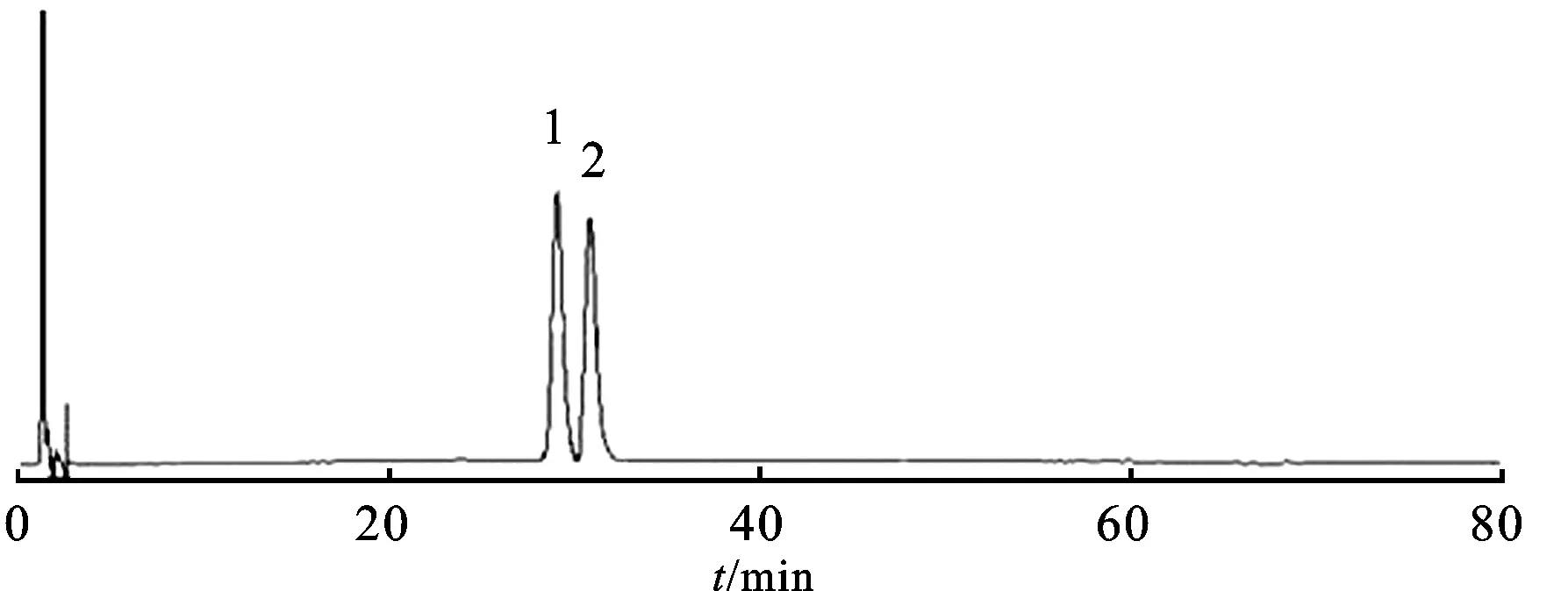
图1系统适用性色谱图
1.杂质2;2.阿托伐他汀钙
Fig.1Systemsuitabilitychromatograms
1.impurity2;2.atorvastatincalcium
2.3专属性为了考证色谱系统是否适用于本品各种降解产物的分离,采用酸、碱、光照、氧化和加热方法对样品溶液进行破坏实验,使其产生降解产物的峰,在HPLC条件下检测杂质峰与主峰分离的情况。(1)酸破坏实验:取本品细粉0.15g,置于20mL量瓶中,加入混合溶剂10mL使其溶解,再加入1mol·L-1的盐酸溶液1.0mL,放置24h,用1mol·L-1的氢氧化钠溶液调至中性;(2)碱破坏实验:取本品细粉0.15g,置于20mL量瓶中,加入混合溶剂10mL溶解,再加入1.0mL1mol·L-1的氢氧化钠溶液,放置24h,用1mol·L-1的盐酸溶液调至中性;(3)氧化破坏:取本品细粉0.15g,置于20mL量瓶中,加入混合溶剂10mL溶解,再加入0.1mL的双氧水(浓度为30mL·L-1),放置12h,加溶剂定容至刻度;(4)光破坏实验:取本品细粉0.15g,置于20mL量瓶中,加入溶剂溶解并定容至刻度,在4 500±500lx的光照箱中放置24h;(5)热破坏实验:取本品细粉0.15g,置于20mL量瓶中,加溶剂溶解并定容至刻度,置于干燥箱中70 ℃放置24h,取出,放至室温。取上述破坏实验溶液,按照2.1 项下条件进行检测,色谱见图2~3。结果显示,主成分峰与酸、碱、光、热和氧化破坏产生的降解物均可达到有效分离,说明该色谱条件可有效分离杂质,且辅料对测定不产生干扰[10]。
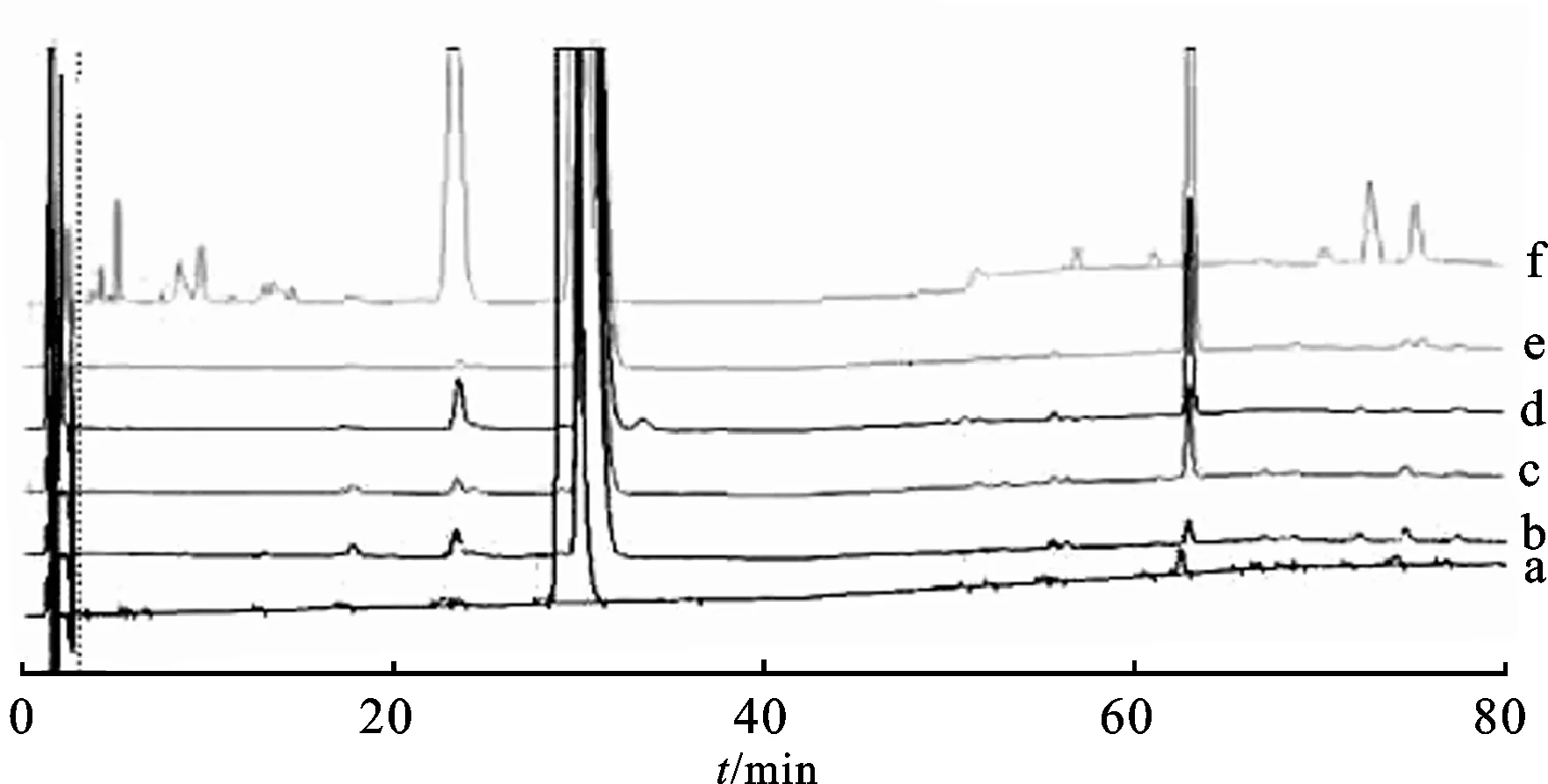
图2 破坏性实验色谱图
a.供试品;b.光照破坏;c.高温破坏;d.碱破坏;e.酸破坏;f.氧化破坏
Fig.2Destructivetestchromatograms
a.testsampleb.destroyedbylight;c.destroyedbyheat;d.destroyedbyalkali;e.destroyedbyacid;f.destroyedbyoxidative
2.4线性及范围分别取阿托伐他汀钙对照品及阿托伐他汀钙杂质1,2,3,4和6对照品适量,按照对照品溶液的制备方法配制成1mL约含阿托伐他汀及阿托伐他汀钙杂质(1,2,3,4 和6) 0.3,0.6,1.2,1.8,2.4和3.0μg的混合溶液(阿托伐他汀钙杂质1,2和3以游离形式即去氟阿托伐他汀、反式阿托伐他汀和双氟阿托伐他汀计算),分别精密量取20μL,注入液相色谱仪,记录色谱图及峰面积积分值。以质量浓度为横坐标(X),以峰面积积分值为纵坐标(Y),对测定结果进行回归分析,杂质1,2,3,4和6的回归方程如下:Y=0.411 2 X-0.001 1,r=0.999 8;Y=0.383 4 X+0.002 8,r=0.999 8;Y=0.390 2 X-0.005 6,r=0.999 8;Y=0.313 7 X-0.008 8,r=0.999 8;Y=0.405 X+0.004 6,r=0.999 8。
结果表明,阿托伐他汀钙杂质1、杂质2、杂质3、杂质4和杂质6的进样量分别在6.182~74.184ng,5.802~69.624ng,6.196~74.352ng,6.0~72.0ng和6.4~64.0ng之间时,各杂质的进样量与峰面积积分值之间呈良好的线性关系。
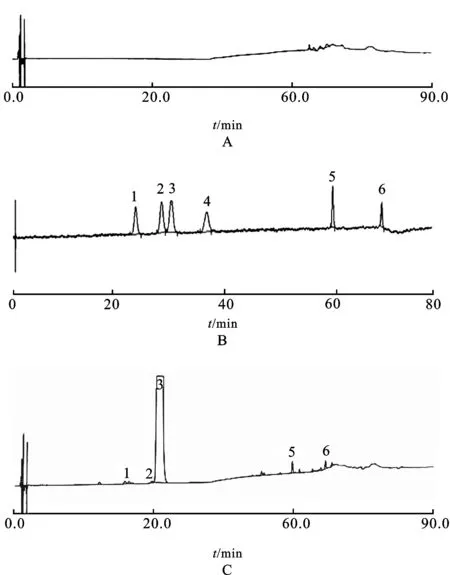
图3HPLC图
A.空白;B.混合对照品;C.供试品;1.杂质1;2.杂质2;3.阿托伐他汀钙;4.杂质3;5.杂质4;6.杂质6
Fig.3HPLCchromatograms
A.blank;B.mixedreferencesubstance;C.testsamples;1.impurity1;2.impurity2;3.atorvastatincalcium;4.impurity3;5.impurity4;6.impurity6
2.5检测限精密量取混合阿托伐他汀及各杂质的混合对照品,用混合溶剂乙腈-水(50∶50)逐步稀释至各主峰的响应值约为噪音水平的3倍时,记录色谱图,得出各检测限分别约为:阿托伐他汀2ng,杂质1 2ng、杂质2 2ng、杂质3 2ng、杂质4 2ng、杂质6 1.6ng。
2.6重复性实验按照供试品溶液制备方法制备供试品溶液6份,吸取20μL,在上述色谱条件下进行测定,记录色谱图和数据。实验结果表明,除总杂质的RSD值为1.87%,其他已知单个杂质测定结果无差异,重复性良好,误差在允许的范围内。
2.7溶液稳定性取供试品溶液和自身对照溶液,分别在0,4,8,26和30h取样20μL,注入液相色谱仪(暗处放置),记录色谱图及峰面积积分值,结果表明,样品溶液中主成分及杂质峰面积在8h内稳定,有关物质个数及量不变,8h后杂质4和杂质6及氧化主杂质变化显著,因此本品有关物质测定供试品溶液应现制,放置不得超过8h。
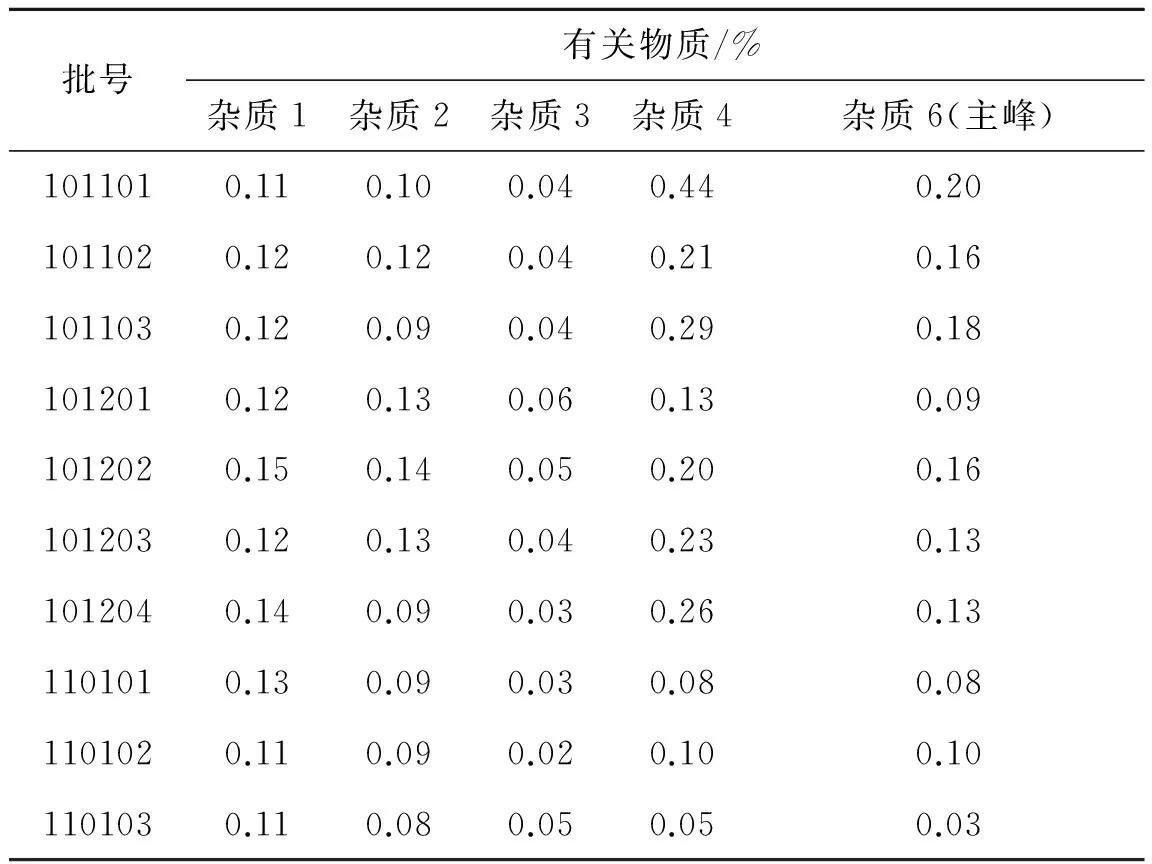
2.8样品测定按照建立的方法测定10批样品,结果见表2。
表210批样品有关物质测定结果(外标法测定)
Tab.2 10batchesofsamplesrelatedsubstancesdeterminationresults(externalstandardmethod)
批号有关物质/%杂质1杂质2杂质3杂质4杂质6(主峰)1011010.110.100.040.440.201011020.120.120.040.210.161011030.120.090.040.290.181012010.120.130.060.130.091012020.150.140.050.200.161012030.120.130.040.230.131012040.140.090.030.260.131101010.130.090.030.080.081101020.110.090.020.100.101101030.110.080.050.050.03
3 讨论
3.1流动相的选择与分析原料标准[YBH00262010]有关物质梯度洗脱,随着四氢呋喃增加基线逐渐明显向上漂移严重,采用扣除基线的方法测定,但色谱峰仍难以辨识且积分不准确,四氢呋喃用量较大、价格昂贵、气味毒性较大。标准[H20030118]方法阿托伐他汀杂质在前40 min分离较好,各个杂质基本达到基线分离,但在75 min后杂质仍不能完全洗脱出柱。欧洲药典和美国药典方法阿托伐他汀杂质在70 min分离较好,各个杂质基本达到基线分离,75 min杂质基本洗脱出柱,但70 min以后基线不平,仍有馒头峰出现。经参考欧洲药典和美国药典方法调整流动相,A相微调,保证阿托伐他汀保留时间在27~34 min之间,运行40 min;调整B相乙腈的比例及梯度时间,确定最终流动相。在调节流动相的过程中,增加乙腈比例和pH值,可使阿托伐他汀保留时间缩短,反之亦然。
在此色谱条件下,阿托伐他汀保留时间在27~34min之间,已知阿托伐他汀钙杂质1,2,3,4和6均能达到基线分离,杂质2与阿托伐他汀分离度≥1.5,其他杂质峰也基本能达到基线分离,杂质在80min能完全被洗脱出柱,基线平稳,继续洗脱至120min无杂质峰出现。
3.2检测波长的选择将阿托伐他汀钙及阿托伐他汀杂质1,2,3,4和6对照品溶液及空白辅料溶液在200~400nm处进行扫描,结果显示,在244和260nm处各溶液所出峰的个数相同,各个峰在260nm下的峰面积约为244nm下的80%~90%。260nm下梯度运行基线比244nm下平稳,更利于色谱峰的积分和判断。
3.3色谱柱及pH值的影响本品有关物质多,因此对色谱柱的要求较高,在已对比使用的色谱柱中以Thermo公司的SyncronisC18(250mm×4.6mm),(填料参数:碳载量16%,粒径5μm,孔径100A,比表面积320m2·g-1,USP分类为L1)分离效果最好。本色谱系统乙腈和pH值对主峰的保留时间影响较大,增加乙腈和升高pH值均会使主峰保留时间缩短,反之亦然。实验以精密pH值计调节pH值在5.0±0.1范围内,必要时调节乙腈比例使阿托伐他汀保留时间在26~34min之间,相对保留时间变化不明显。
[1]张亚平,杨淑莲,陈芬儿.HPLC法检测阿托伐他汀钙有关物质的含量及光学纯度[J].药物分析杂志,2010,30(12):2311-2313.
[2]李文莉,钟庆元,文庆.HPLC法测定阿托伐他汀钙胶囊的含量及有关物质[J].药物分析杂志,2007,27(2):267-269.
[3]王艳丽,于晓原,张璐璐,等.阿托伐他汀钙治疗老年冠心病合并高脂血症的疗效观察[J].西北药学杂志,2013,28(6):630-631.
[4]汪钦标,杨成钰.HPLC法测定阿托伐他汀钠含量及有关物质[J].轻工科技,2012,(3):124-125.
[5]蒋厦,沈卫阳.RP-HPLC法测定阿托伐他汀钙片的有关物质[J].海峡药学,2013,25(8):60-62.
[6]WS1-(X-106)-2003Z,国家食品药品监督管理局标准[S].
[7]YBH00262010,国家食品药品监督管理局标准[S].
[8]USP34-29NF29 [S].2011:1949.
[9]EP7.1[S].2011:3380.
[10]王嫦鹤,焦艳,杜珊,等.HPLC法测定乌拉地尔不同制剂中的有关物质[J] 西北药学杂志,2013,28(3): 254-257.
Determination of the related substances in Atorvastatin Calcium Tablets by HPLC
YU Nina1,ZHANG Ling2,MENG Shuhua2
(1.ShaanxiProvincialCenterforNewDrugEvaluation,Xi′an710065,China;2Xi′anTianyiqinkunPharmaceuticalLimitedCompany,Xi′an710077,China)
ObjectiveToestablishamethodforthedeterminationoftherelatedsubstancesinAtorvastatinCalciumTablets.MethodsRP-HPLCwasadoptedtodeterminethecontentofatorvastatincalcium.ThedeterminationwasperformedonaSyncronisC18column(250mm×4.6mm,5μm)withthemobilephaseAconsisitedofacetatebuffer-acetonitrile-tetrahydrofuran(61∶27∶12),andthemobilephaseBconsisitedofacetatebuffer-acetonitrile-tetrahydrofuran(47∶41∶12).Theflowratewas1.5mL·min-1,andthedetectivewavelengthwassetat260nm.Thecolumntemperaturewas35 ℃.ResultsTheimpurities1,2,3,4,6andatorvastatincalciumpeakswerebaselinelyseparated.Thelimitofdetectionofatorvastatinwas2ng,andthelimitsofimpurities1,2,3,4and6were2,2,2,2and1.6ng.Therepeatbilityperformedwell,andtheRSDwas1.87%(n=6).ConclusionThemethodisreliable,stable,andspecific,anditissuitableforthethedeterminationoftherelatedsubstancesinAtorvastatinCalciumTablets.
atorvastatincalcium;relatedsubstances;determinationofimpurities;HPLC
10.3969/j.issn.1004-2407.2016.05.008
R917
A
1004-2407(2016)05-0466-04
2015-12-01)
猜你喜欢
医学食疗与健康(2022年2期)2022-04-23
中华养生保健(2020年8期)2021-01-14
科学(2020年5期)2020-11-26
中国惯性技术学报(2019年3期)2019-10-15
环境与生活(2019年1期)2019-09-10
中成药(2017年10期)2017-11-16
舰船电子对抗(2016年5期)2016-12-13
中国继续医学教育(2015年6期)2016-01-07
中国当代医药(2015年16期)2015-03-01
中国卫生标准管理(2015年17期)2015-01-26
