
创新霉素前药衍生物的合成及抗菌活性研究
2016-09-05 08:45刘宗英朱俊泰金洁李卓荣
中国医药生物技术 2016年4期
刘宗英,朱俊泰,金洁,李卓荣
·论著·
创新霉素前药衍生物的合成及抗菌活性研究
刘宗英,朱俊泰,金洁,李卓荣
目的合成创新霉素前药衍生物,测定其体外抗菌活性。
方法以创新霉素为起始原料,经酯化或酰胺化反应得到目标化合物 1a ~ f 和 2a ~ f。
结果共合成 12 个创新霉素前药衍生物,其结构均经核磁共振氢谱和质谱确证;化合物 1a ~ e 体现了较强的体外抗甲氧西林敏感金葡菌(MSSA)、耐甲氧西林金葡菌(MRSA)、甲氧西林敏感表葡菌(MSSE)、耐甲氧西林表葡菌(MRSE)和肺炎链球菌(ATCC49619)活性,其中化合物 1a、1e 体外抗 MSSA、MRSA、MSSE、MRSE、ATCC49619 活性是创新霉素的 4 ~ 128 倍,相当或略优于阳性对照药替甲环素。
结论在创新霉素的 C-2 羧酸上形成取代苯甲酰氧甲酯或环己基甲酰氧甲酯前药有利于提高体外抗革兰阳性菌活性,化合物 1a、1e 的体内抗菌活性值得进一步研究。
化学技术,合成; 药物前体;抗菌活性;创新霉素
www.cmbp.net.cn中国医药生物技术, 2016, 11(4):314-318
随着抗生素的广泛使用,一些菌株如革兰阳性菌中的耐甲氧西林金葡菌(MRSA)、耐青霉素肺炎链球菌(PRSP)和耐万古霉素肠球菌(VRE),革兰阴性菌中的铜绿假单胞菌和鲍曼不动杆菌开始出现多药耐药性。由于当前细菌耐药形势严峻,尤其超级耐药性细菌的出现,使研发新的抗耐药菌新药迫在眉睫[1-4]。
创新霉素[5]是由山东济南土壤中分离出的一株游动放线菌(Actinoplanes tsinanensis)发酵产生,是我国发现的第一个全新结构骨架抗生素。创新霉素具有中等抗菌谱,对部分革兰阳性与阴性菌有抑制作用且毒性很低。小鼠体内活性研究结果表明,创新霉素对金葡菌、痢疾杆菌和大肠杆菌感染的小鼠均具有较强的治疗作用。创新霉素在临床上对大肠杆菌引起的败血症、泌尿系统感染、肠道感染等有较好疗效。由于创新霉素具有中等抗菌谱,影响其在临床上的广泛使用。本文以创新霉素为先导化合物,设计合成了一系列创新霉素前药衍生物并测定其体外抗菌活性,以期发现抗菌活性增强,药代动力学特性更优的新型抗菌药物。
1 材料与方法
1.1材料
1.1.1主要仪器A400 多点接种仪为英国Denley 公司产品;MP 90 熔点仪为瑞士 Mettler Toledo 公司产品,温度计未经校正;Bruker 400 ultrashieldTM核磁仪为德国 Bruker 公司产品;70-SE 质谱仪为英国 VG 公司产品。
1.1.2主要试剂体外测活所用创新霉素为自制(纯度 > 99.1%),其余试剂均为市售分析纯或化学纯。
1.2方法
1.2.1目标化合物的合成
1.2.1.1(2R,3S)-3-甲基-3,5-2H-噻喃[4,3,2-cd]吲哚-2-羧酸酯的合成以创新霉素为起始原料,在三乙胺作用下与取代甲酸氯甲酯(5a ~ f)反应得到目标化合物 1a ~ f[6-7]。其中,起始原料创新霉素为微生物发酵产物;前药修饰物 1a ~ f 制备中所需原料5a ~ f 的制备以取代甲酰氯为起始原料,在氯化锌作用下与多聚甲醛发生酯化反应制得[8](图 1)。
1.2.1.2(2R,3S)-3-甲基-3,5-2H-噻喃[4,3,2-cd]吲哚-2-羧酸酯/酰胺的合成以创新霉素为起始原料,在三乙胺作用下与取代醇或取代氨(6a ~ f)发生酯化或酰胺化反应得到目标化合物 2a ~ f[9](图 2)。
1.2.2体外抗菌活性采用美国国家临床实验室标准化委员会(NCCLs)推荐的平皿二倍稀释法测定目标化合物 1a ~ f、2a ~ f 的最低抑菌浓度(MIC),阳性对照药为创新霉素和替甲环素。于无菌平皿内加入 1 ml 药液,再加入融化的 50 ℃ MH培养基 14 ml,混匀,使其每皿内所含药物终浓度依次为 128、64、32、16、8、4、2、1、0.5、0.25、0.125、0.06、0.03 mg/L;待冷却后用多点接种仪接种细菌,接种菌量约为 105CFU/ml。置于 35 ~37 ℃ 培养箱内培养 24 h,观察记录结果,无菌生长的平皿中所含药物最小的浓度即为最低抑菌浓度。测定菌株分别为甲氧西林敏感金葡菌(MSSA)、耐甲氧西林金葡菌(MRSA)、甲氧西林敏感表葡菌(MSSE)、耐甲氧西林表葡菌(MRSE)、肺炎克雷伯菌(E+和 E-)、大肠埃希菌(E+和 E-,ATCC25922)、铜绿假单胞菌、金黄色葡萄球菌(ATCC25923)和肺炎链球菌(ATCC49619)。其中大肠埃希菌 E+和肺炎克雷伯菌 E+为产超广谱 β-内酰胺酶菌株。
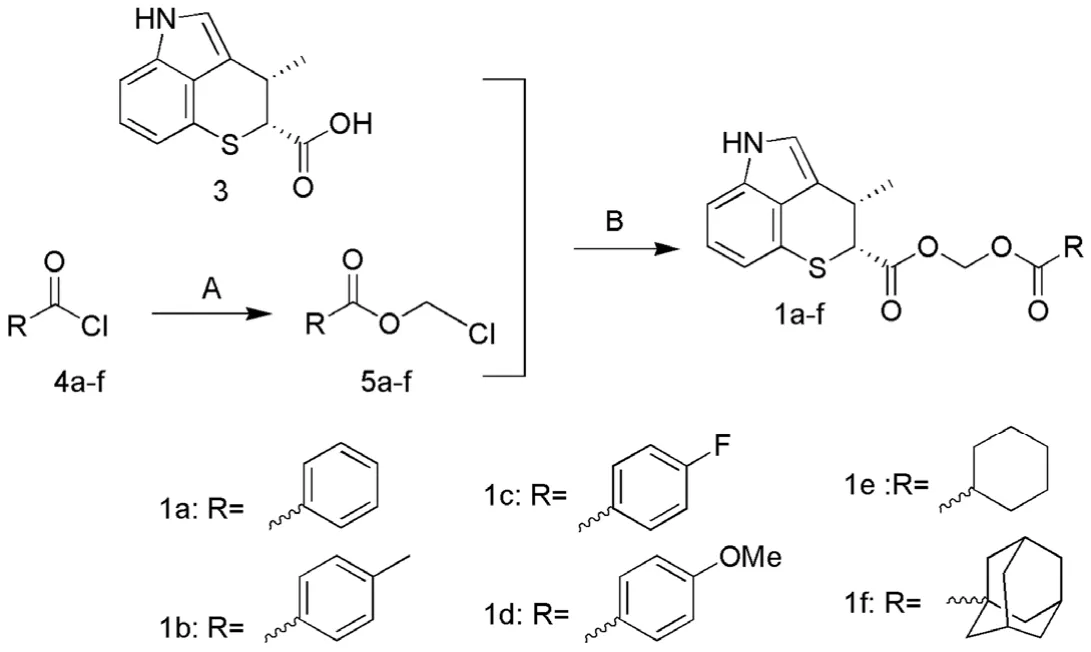
图1 化合物 1a ~ f 的合成路线Figure 1 Synthesis of compounds 1a - f

图2 化合物 2a ~ f 的合成路线Figure 2 Synthesis of compounds 2a - f
2 结果
2.1目标化合物的合成
共合成 12 个创新霉素前药衍生物,所有化合物的结构均经过1H-NMR 和 MS 分析确证。
2.1.1(2R,3S)-3-甲基-3,5-2H-噻喃[4,3,2-cd]吲哚-2-羧酸(苯甲酰氧)甲酯(1a)淡黄色固体,收率 59%,mp:116 ~ 118 ℃。1H-NMR(400 MHz,DMSO-d6)δ 10.92(s,1H),7.95(d,J = 7.2 Hz,2H),7.21(t,J = 6.0 Hz,1H),7.57(t,J = 8.0 Hz,2H),7.17(d,J = 1.6 Hz,1H),7.12(d,J = 8.0 Hz,1H),6.99(d,J = 7.2 Hz,1H),6.77(d,J = 7.2 Hz,1H),6.02(d,J = 6.0 Hz,1H),5.92(d,J = 6.0 Hz,1H),4.42(d,J = 4.0 Hz,1H),3.2(m,1H),1.23(d,J = 6.8 Hz,3H)。MS(ESI+)m/z:390.1 [M+ Na]+。
2.1.2(2R,3S)-3-甲基-3,5-2H-噻喃[4,3,2-cd]吲哚-2-羧酸(4'-甲基苯甲酰氧)甲酯(1b)淡黄色固体,收率 52%,mp:140 ~ 143 ℃。1H-NMR(400 MHz,DMSO-d6)δ 10.92(s, 1H),7.84(d,J = 8.0 Hz,2H),7.37(d,J = 8.0 Hz,2H),7.16 (d,J = 1.6 Hz,1H),7.12(d,J = 8.0 Hz,1H),7.00(t,J = 7.6 Hz,1H),6.77(d,J = 7.2 Hz,1H),6.00(d,J = 6.0 Hz,1H),5.92(d,J = 6.0 Hz,1H),4.42(d,J = 3.6 Hz,1H),3.62(m,1H),2.40(s,3H),1.22(d,J = 6.8 Hz,3H)。MS(ESI+)m/z:404.1 [M+ Na]+。
2.1.3(2R,3S)-3-甲基-3,5-2H-噻喃[4,3,2-cd]吲哚-2-羧酸(4'-氟苯甲酰氧)甲酯(1c)淡黄色固体,收率 54%,mp:57 ~ 60 ℃。1H-NMR(400 MHz,DMSO-d6)δ 10.92(s,1H),7.99(d,J = 8.0 Hz,2H),7.38(d,J = 8.0 Hz,2H),7.16(s,1H),7.12(d,J = 8.0 Hz,1H),7.00(t,J = 8.0 Hz,1H),6.77(d,J = 7.2 Hz,1H),6.00(d,J = 6.0 Hz,1H),5.93(d,J = 6.0 Hz,1H),4.42(d,J = 3.6 Hz,1H),3.62(m,1H),1.24(d,J = 6.8 Hz,3H)。MS(ESI+)m/z:408.1 [M+ Na]+。
2.1.4(2R,3S)-3-甲基-3,5-2H-噻喃[4,3,2-cd]吲哚-2-羧酸(4'-甲氧基苯甲酰氧)甲酯(1d)淡黄色固体,收率 37%,mp:118 ~ 119 ℃。1H-NMR (400 MHz,DMSO-d6)δ 10.92(s,1H),7.91(d,J = 8.8 Hz,2H),7.16(d,J = 1.6 Hz,1H),7.12 (d,J = 8.4 Hz,1H),7.08(d,J = 8.4 Hz,2H)7.00(t,J = 7.6 Hz,1H),6.77(d,J = 7.6 Hz,1H),5.98(d,J = 6.0 Hz,1H),5.91(d,J = 6.0 Hz,1H),4.42(d,J = 4.0 Hz,1H),3.85(s,3H)3.62 (m,1H),1.22(d,J = 6.8 Hz,3H)。MS(ESI+)m/z:420.1 [M+ Na]+。
2.1.5(2R,3S)-3-甲基-3,5-2H-噻喃[4,3,2-cd]吲哚-2-羧酸(环己基甲酰氧)甲酯(1e)淡黄色固体,收率 58%,mp:99 ~ 101 ℃。1H-NMR(400 MHz,DMSO-d6)δ 10.93(s,1H),7.18(s,1H)7.13 (d,J = 8.0 Hz,1H)7.00(t,J = 8.0 Hz,1H),6.79(d,J = 7.6 Hz,1H),5.73(d,J = 6.0 Hz,1H),5.68(d,J = 6.0 Hz,1H),4.35(d,J = 3.6 Hz,1H),3.59(m,1H),2.31(m,1H),1.79(m,2H),1.67(m,2H),1.57(m,1H),1.26 (m,8H)。MS(ESI+)m/z:396.1 [M+ Na]+。
2.1.6(2R,3S)-3-甲基-3,5-2H-噻喃[4,3,2-cd]吲哚-2-羧酸(金刚烷甲酰氧)甲酯(1f)淡黄色固体,收率 69%,mp:147 ~ 150 ℃。1H-NMR (400 MHz,DMSO-d6)δ 10.93(s,1H),7.18(d,J = 1.2 Hz,1H),7.13(d,J = 8.0 Hz,1H),7.00(t,J = 7.6 Hz,1H),6.77(d,J = 7.2 Hz,1H),5.73(d,J = 5.6 Hz,1H),5.67(d,J = 5.6 Hz,1H),4.34(d,J = 4.0 Hz,1H),3.58(m,1H),1.97(s,3H),1.77(d,J = 2.4 Hz,6H),1.66(m,6H),1.27(d,J = 6.8 Hz,3H)。MS(ESI+)m/z:448.2 [M+ Na]+。
2.1.7(2R,3S)-3-甲基-3,5-2H-噻喃[4,3,2-cd]吲哚-2-甲酰(2'-吗啉代)乙胺(2a)淡黄色固体,收率 63%,mp:203 ~ 205 ℃。1H-NMR(400 MHz,DMSO-d6)δ 10.88(s,1H),7.87(s,1H),7.15 (d,J = 2.0 Hz1H),7.09(d,J = 8.0 Hz,1H),6.99(t,J = 7.6 Hz,1H),6.76(d,J = 7.2 Hz,1H),4.10(d,J = 3.6 Hz,1H)3.59(m,1H),3.54(t,J = 4.4 Hz,4H),3.29(m,1H),3.11(m,1H),2.32(m,6H),1.21(d,J = 6.8 Hz,3H)。MS(ESI+)m/z:346.2 [M+ H]+。
2.1.8(2R,3S)-3-甲基-3,5-2H-噻喃[4,3,2-cd]吲哚-2-甲酰(2'-吗啉代)乙酯(2b)淡黄色固体,收率 61%,mp:125 ~ 128 ℃。1H-NMR(400 MHz,DMSO-d6)δ 10.91(s,1H),7.17(d,J = 2.0 Hz,1H),7.11(d,J = 8.4 Hz,1H),7.00(t,J = 7.6 Hz,1H),6.78(d,J = 7.2 Hz,1H),4.29(d,J = 3.6 Hz,1H),4.12(m,2H),3.55(m,1H),3.52(m,4H),2.39(m,2H),2.31(m,4H),1.27(d,J = 6.8 Hz,3H)。MS(ESI+)m/z:347.2 [M+ H]+。
2.1.9(2R,3S)-3-甲基-3,5-2H-噻喃[4,3,2-cd]吲哚-2-甲酰(2'-二甲氨基)乙胺(2c)淡黄色固体,收率 78%,mp:209 ~ 212 ℃。1H-NMR(400 MHz,DMSO-d6)δ 10.87(s,1H),7.95(s,1H),7.14 (d,J = 2.0 Hz,1H),7.09(d,J = 8.0 Hz,1H),6.98(t,J = 7.6 Hz,1H),6.75(d,J = 7.2 Hz,1H),4.11(d,J = 3.2 Hz,1H),3.58(m,1H),3.22(m,1H),3.09(m,1H),2.26(t,J = 6.8 Hz,2H),2.10(d,J = 3.6 Hz,6H),1.19(d,J = 6.8 Hz,3H)。MS(ESI+)m/z:304.1 [M+ H]+。
2.1.10(2R,3S)-3-甲基-3,5-2H-噻喃[4,3,2-cd]吲哚-2-甲酰(3'-二甲氨基)丙胺(2d)淡黄色固体,收率 66%,mp:169 ~ 170 ℃。1H-NMR(400 MHz,DMSO-d6)δ 10.87(s,1H),8.08(t,J = 4.4 Hz,1H),7.14(d,J = 2.0 Hz,1H),7.08(d,J = 8.0 Hz,1H),6.98(t,J = 7.6 Hz,1H),6.75(d,J = 7.2 Hz,1H),4.11(d,J = 3.6 Hz,1H),3.59(m,1H),3.16(m,1H),3.03(m,1H),2.19(t,J = 7.2 Hz,2H),2.11(s,6H),1.53(m,2H),1.19(d,J =6.8 Hz,3H)。MS(ESI+)m/z:318.2 [M+ H]+。
2.1.11(2R,3S)-3-甲基-3,5-2H-噻喃[4,3,2-cd]吲哚-2-甲酰(3'-咪唑)丙胺(2e)淡黄色固体,收率 47%,mp:218 ~ 220 ℃。1H-NMR(400 MHz,DMSO-d6)δ 10.88(s,1H),8.19(t,J = 5.2 Hz,1H),7.60(s,1H),7.15(t,J = 5.2 Hz,2H),7.09(d,J = 8.0 Hz,1H),6.98(t,J = 8.0 Hz,1H),6.89(s,1H),6.76(d,J = 7.2 Hz,1H),4.14(d,J = 3.6 Hz,1H),3.96(t,J = 7.2 Hz,2H),3.63 (m,1H),3.09(m,1H),3.01(m,1H),1.84 (m,2H),1.19(d,J = 6.8 Hz,3H)。MS(ESI+)m/z:341.1 [M+ H]+。
2.1.12(2R,3S)-3-甲基-3,5-2H-噻喃[4,3,2-cd]吲哚-2-甲酰(3'-氧代吡咯烷)丙胺(2f)淡黄色固体,收率 56%,mp:183 ~ 184 ℃。1H-NMR (400 MHz,DMSO-d6)δ 10.87(s,1H),8.13(t, J = 5.2 Hz,1H),7.14(s,1H),7.08(d,J = 8.0 Hz,1H),6.98(t,J = 7.6 Hz,1H),6.75(d,J = 7.2 Hz,1H),4.12(d,J = 3.6 Hz,1H),3.61(m,1H),3.23(m,3H),3.06(m,1H),2.20(m,2H),1.92(m,2H),1.57(m,2H),1.18(d,J = 6.8 Hz,3H)。MS(ESI+)m/z:358.2 [M+ H]+。
2.2体外抗菌活性
采用平皿二倍稀释法测定了目标化合物体外抗菌活性,其最小抑菌浓度(MIC)结果如表 1 所示。研究结果表明化合物 1a ~ e 显示了较强的体外抗 MSSA、MRSA、MSSE、MRSE 和 ATCC49619活性,MIC 值为 0.125 ~ 16 µg/ml;化合物 2b、2c、2f 显示了较弱的抗 MSSA 活性(MIC 值为 4 ~64 μg/ml);化合物 1a - f、2a - d、2f 显示了较弱的抗铜绿假单胞菌活性(MIC 值为 16 ~ 64 μg/ml)。
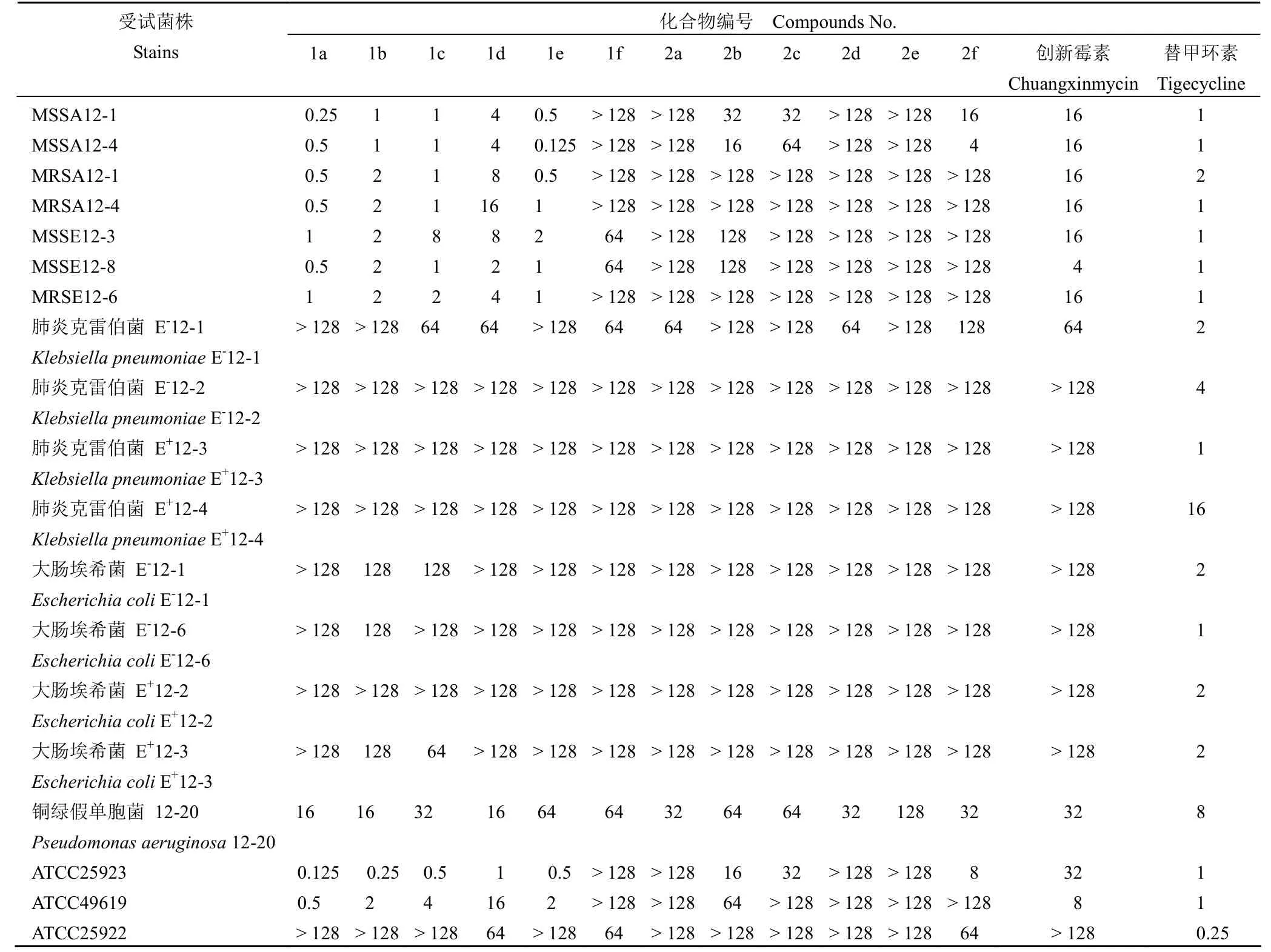
表1 化合物 1a ~ f、2a ~ f 的体外最小抑菌浓度(μg/ml)Table 1 In vitro antibacterial activities of 1a - f, 2a - f (μg/ml)
3 讨论
目标化合物体外抗菌活性结果表明化合物1a ~ e 体外抗 MSSA、MRSA、MSSE、MRSE 和ATCC49619 活性强于先导化合物创新霉素,其中化合物 1a、1e 体外抗 MSSA、MRSA、MSSE、MRSE、ATCC49619 活性是创新霉素的 4 ~ 128 倍,化合物 1a、1e 体外抗 MSSA、MRSA 活性是阳性对照药替甲环素的 1 ~ 8 倍,化合物 1a、1e 体外抗 MSSE、MRSE 活性与替甲环素相当,表明在创新霉素的 C-2 羧酸上形成取代苯甲酰氧甲酯(1a ~d)或环己基甲酰氧甲酯(1e)前药有利于提高创新霉素体外抗革兰阳性菌活性;化合物 1b ~ d 的体外抗 MSSA、MRSA、MSSE、MRSE 和ATCC49619 活性较化合物 1a 下降了 2 ~ 32 倍,说明在 C-2 酯基的苯环上增加取代基团(甲基、氟、甲氧基)不利于抗菌活性提高;化合物 1a ~ f、2a ~ d、2f 体外抗铜绿假单胞菌活性与创新霉素相当,但弱于阳性对照药替甲环素(MIC 值为8 μg/ml),说明在创新霉素的 C-2 羧酸上形成酯/酰胺前药未能提高抗革兰阴性菌活性。化合物 1a、1e 的体内抗菌活性值得进一步研究。
[1] Cheng G, Dai M, Ahmed S, et al. Antimicrobial drugs in fighting against antimicrobial resistance. Front Microbiol, 2016, 7:470.
[2] Rossolini GM, Arena F, Pecile P, et al. Update on the antibiotic resistance crisis. Curr Opin Pharmacol, 2014, 18:56-60.
[3] Lee HH, Molla MN, Cantor CR, et al. Bacterial charity work leads to population-wide resistance. Nature, 2010, 467(7311):82-85.
[4] Willems RJ, Hanage WP, Bessen DE, et al. Population biology of Gram-positive pathogens: high-risk clones for dissemination of antibiotic resistance. FEMS Microbiol Rev, 2011, 35(5):872-900.
[5] Wang YC. Review of developments in chuangxinmycin. Chin Pharm J,1992, 27(9):520-522. (in Chinese)
王玉成. 创新霉素研究概况. 中国药学杂志, 1992, 27(9):520-522.
[6] Singh SB, Rindgen D, Bradley P, et al. Design, synthesis,structure-function relationship, bioconversion, and pharmacokinetic evaluation of ertapenem prodrugs. J Med Chem, 2014, 57(20):8421-8444.
[7] Gupta D, Varghese Gupta S, Dahan A, et al. Increasing oral absorption of polar neuraminidase inhibitors: a prodrug transporter approach applied to oseltamivir analogue. Mol Pharm, 2013, 10(2):512-522.
[8] Cai J, Zhou W, Chen JQ, et al. Synthesis and crystal structure of benzoyloxymethyl(4α,8β,13β)-13-methyl-16-oxo-17-norkauran-18-carbonate ester. J Chem Crystallogr, 2009, 39:108-111.
[9] Su SH, Tu JD, Zhang SW. Synthesis of some derivatives of chuangxinmycin. Pharml Ind, 1984(2):17-21. (in Chinese)
苏盛惠, 屠健德, 张士纬. 创新霉素衍生物的合成. 医药工业,1984(2):17-21.
【Abstract】
ObjectiveTo synthesize and evaluate the in vitro antibacterial activities of Chuangxinmycin prodrugs.
MethodsCompounds 1a - f and 2a - f were synthesized via esterification or amidation of Chuangxinmycin.
ResultsThe structure of 12 Chuangxinmycin prodrugs synthesized in this paper was confirmed by1H-NMR and MS. Compounds 1a - 1e showed potent antibacterial activity against MSSA, MRSA, MSSE, MRSE and ATCC49619. The antibacterial activities of compounds 1a, 1e against MSSA, MRSA, MSSE, MRSE,ATCC49619 were 4 to 128-fold stronger than that of Chuangxinmycin,and similar to or stronger than that of Tigecycline.
ConclusionProdrugs with a C-2 carboxyl benzoyloxy methyl ester or cyclohexanecarboxyloyloxy methyl ester of Chuangxinmycin are favorable for increasing the in vitro antibacterial activities on Gram-positive bacteria. The in vivo antibacterial activities of compounds 1a and 1e deserve further investigation.
Author Affiliations: Center for Drug Evaluation, CFDA, Beijing 100038, China (LIU Zong-ying);Department of Medicinal Chemistry, Institute of Medicinal Biotechnology, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing 100050, China (LIU Zong-ying, ZHU Jun-tai, JIN Jie, LI Zhuo-rong)
www.cmbp.net.cnChin Med Biotechnol, 2016, 11(4):314-318
Synthesis and antibacterial activity evaluation of Chuangxinmycin prodrugs
LIU Zong-ying, ZHU Jun-tai, JIN Jie, LI Zhuo-rong
Chemistry techniques, synthesis;Prodrugs;Antibacterial activity;Chuangxinmycin
LI Zhuo-rong, Email: l-z-r@263.net
10.3969/j.issn.1673-713X.2016.04.005
国家自然科学基金(81373268);“重大新药创制”国家科技重大专项(2014ZX09507-009-04)
100038 北京,国家食品药品监督管理总局药品审评中心(刘宗英);100050 北京,中国医学科学院北京协和医学院医药生物技术研究所合成室(刘宗英、朱俊泰、金洁、李卓荣)
李卓荣,Email:l-z-r@263.net
2016-05-11
猜你喜欢
中国油脂(2022年1期)2022-02-12
中国饲料(2021年17期)2021-11-02
山东第一医科大学(山东省医学科学院)学报(2021年7期)2021-10-13
烟台大学学报(自然科学与工程版)(2021年1期)2021-03-19
昆明医科大学学报(2020年12期)2021-01-26
化工时刊(2020年7期)2020-09-04
化工技术与开发(2020年8期)2020-08-26
河南化工(2020年4期)2020-06-04
人物画报(2020年36期)2020-03-13
中成药(2018年5期)2018-06-06
