
急性缺糖缺氧通过增强胆碱酯酶表达促进肾小管细胞的炎性损伤
2016-09-03 07:19吴明吴乐锋李明利陆俊福赖凯徐迹刘芬冯永文
实验与检验医学 2016年4期
吴明,吴乐锋,李明利,陆俊福,赖凯,徐迹,刘芬,冯永文
(1、深圳市第二人民医院重症医学科,广东 深圳 5108035;2、深圳市第二人民医院介入治疗科,广东 深圳 5108035;3、深圳市第二人民医院中心实验室,广东 深圳5108035;4、南昌大学第一附属医院重症医学科,江西 南昌330006)
·论著·
急性缺糖缺氧通过增强胆碱酯酶表达促进肾小管细胞的炎性损伤
吴明1,吴乐锋1,李明利2,陆俊福1,赖凯1,徐迹3,刘芬4,冯永文1
(1、深圳市第二人民医院重症医学科,广东深圳5108035;2、深圳市第二人民医院介入治疗科,广东深圳5108035;3、深圳市第二人民医院中心实验室,广东深圳5108035;4、南昌大学第一附属医院重症医学科,江西南昌330006)
目的探讨急性缺糖缺氧导致肾小管细胞损伤的机制。方法分离培养大鼠肾内巨噬细胞、肾小管上皮细胞,构建两者共培养(transwell)模型,细胞分成对照组及缺糖缺氧(Oxygen and glucose deprivation,OGD)组,给予缺糖缺氧处理细胞1h后再正常培养24h,ELISA法检测两组上清液TNF-α,IL-1β和IL-10的浓度,噻唑蓝(MTT)检测肾小管细胞活力及RT-qPCR及Western Blot检测AChE的mRNA和蛋白的表达。结果在共培养上清液中,对照组与OGD相比,TNF-α(pg/ml):(231.67±36.28)VS(428.67±43.16)(P<0.05),IL-1β(pg/ml):(116.67±21.64)VS(219.63±43.86)(P<0.05),IL-10(pg/ml):(235.67± 39.35)VS(432.67±49.72)(P<0.01)。肾小管细胞活力明显降低,分别为(88.41±18.25)VS(46.98±13.87)(P<0.01),OGD组巨噬细胞AChE mRNA和蛋白水平均高于对照组,分别上调了(3.82±0.73)和(2.17±0.46)倍(P<0.01)。结论急性缺糖缺氧增强了肾脏巨噬细胞胆碱酯酶的表达,通过炎症介质,介导了急性缺糖缺氧性肾小管细胞损伤。
缺糖缺氧;胆碱酯酶;炎症介质;肾小管细胞活力
在急性缺氧等各种应激下,毛细血管内皮细胞损伤,白细胞聚集、黏附、激活血小板,释放大量的炎症介质,直接介导了肾脏间质内的巨噬细胞的微环境发生改变,使足细胞逐渐向纤维细胞分化,促进了肾脏纤维化的发生[1]。研究显示急性肾损伤(AKI)的发生除全身血流动力学改变因素外,还与肾脏微环境和炎症、免疫密切相关[2]。AKI是慢性肾脏疾病(CKD)、终末期肾病(ESRD)、死亡和其他非肾脏疾病的独立危险因素[3]。因此,全面理解肾脏微环境、免疫、炎症的关系,对急性肾脏疾病的治疗至关重要[4]。
神经-免疫的正负反馈精准调节,对稳定人类健康至关重要。研究显示胆碱能抗炎通路是由迷走神经介导的实时调控炎症反应通路[5]。当机体发生炎症反应时,局部炎症因子刺激迷走传入神经纤维,将炎症信号传入下丘脑,再经迷走传出神经纤维向炎症部位释放乙酰胆碱(acetylcholine,ACh),激活单核-巨噬细胞上的烟碱型乙酰胆碱受体7(nicotinic acetylcholine receptor alpha7,nACh-Rα7),从而抑制单核-巨噬细胞的活化,减少TNF-α等促炎因子的释放,达到调控炎症反应的目的[6]。神经元细胞分泌的乙酰胆碱酯酶(acetylcholinesterse,AChE)对乙酰胆碱的分解起重要作用。乙酰胆碱酯酶(acetylcholinesterse,AChE)不仅存在于神经元细胞中,也存在于免疫细胞上,其中巨噬细胞表达最丰富[7]。因此,研究急性缺糖缺氧性肾脏疾病的神经-免疫调节机制,对急性缺糖缺氧性肾损伤的治疗提供新方向。为此,我们先通过分离培养肾脏内的巨噬细胞与肾小管上皮细胞,构建两者共培养(transwell)模型,探讨缺糖缺氧对肾小管细胞活力的影响,并观察缺糖缺氧对肾脏巨噬细胞胆碱酯酶的影响。
1 材料与方法
1.1肾脏内巨噬细胞分离与培养脱颈处死SD大鼠(8W,SFP级,湖南斯莱克景达实验动物有限公司),75%酒精侵泡5min,将大鼠倒立提起,外科方法无菌摘取双侧肾脏,置于盛有预冷RPMI-1640培养液(含1%小牛血清)(Gibco美国)的平皿中,剪去结缔组织和脂肪。挤压肾组织通过200目的钢丝网,获得单个细胞。离心200×g10min,去上清液。用含1%小牛血清的RPMI-1640培养液将细胞配成约1×108/ml。获取单个细胞接种于10%FBS 的DMEM培养液(Gibco美国)于细胞培养瓶中,放置37℃、体积分数5%的CO2培养箱(thermo美国)饱和湿度下培养。用贴壁法纯化巨噬细胞并显微镜拍照(Olympus,日本)。将肾内巨噬细胞系按3.0× 104cells/ml的密度接种于24孔板上,每孔1~2ml,培养24h后,将培养液换为无血清培养基,培养24h弃原培养液,并经形态学鉴定,镜下可观察到贴壁肾巨噬细胞的细胞呈圆形、椭圆形、三角形或不规则形,有伪足和突起。将此细胞鉴定为巨噬细胞后作为实验细胞。
1.2肾小管细胞分离与培养动物处理同巨噬细胞分离。分离肾皮质和髓质,取肾皮质剪碎至1mm3大小,使用PBS(Gibco美国)冲洗后离心300×g 10min,反复3次,弃上清液加入配制1g/L I型胶原酶的DMEM消化液,37℃振荡消化30min,并用移液管轻轻吹到30次,将尚未消化的肾皮质更换到新的胶原酶溶液中,继续震荡消化30min,再次吹打30次,将消化后的细胞悬液经100目不锈钢筛网过滤,并用玻璃棒轻轻研磨同时以DMEM冲洗,网下液体再经200目不锈钢筛网过滤后将其转移到离心管中,离心300×g 5min,弃上清液取沉淀,反复2次。使用5ml DEME培养液重悬浮沉淀,并轻轻铺于预先配置好的45%的percoll(GE Healthcare美国)细胞分离液上,使用高速冷冻离心机离心(4000×g,4℃,30min),离心后可见液体分层,吸取近管底第2层细胞悬液即为肾小管上皮细胞。将分离纯化的肾小管上皮细胞用DMEM培养液洗涤并以300×g离心10min,洗涤2次以去残留的Percoll细胞分离液。最后用含10%小牛血清的DMEM培养液悬浮细胞接种于T25细胞培养瓶中并做好标记,台盼蓝(Sigma美国)染色计数细胞并观察细胞活力,将细胞配成所需的浓度,经倒置显微镜(Olympus日本)观察到贴壁细胞体积较大,呈多边鹅卵石样,细胞之间排列紧密,鉴定为肾小管细胞后进行原代培养及传代培养。
1.3缺糖缺氧(OGD)及共培养模型的构建将第3代肾小管上皮细胞以5×103/cm2的密度接种于96孔培养板中,当细胞长满约50%~60%时,10%FBS 的DMEM培养液300μl,放入肾脏巨噬细胞双细胞培养系统中(transwell板)并培养24h。弃原培养液后,正常组细胞:加入Earle's液(Gibco美国);缺糖缺氧组(10%XB+OGD):用无糖Earle's液清洗细胞1次后,分别加入4mmol/ml浓度的Na2S2O4无糖Earle's液,无糖低氧损伤时间为1h。1h后细胞分别用Earle's液清洗1次,加入完全培养液(10% FBS的1640 Gibco美国)培养至24h。
1.4酶联免疫吸附检测按照ELISA试剂盒说明,检测培养细胞的上清液中TNF-α(mybiosource,MBS355371),IL-1β(mybiosource,MBS175941),IL-10(Mybiosource,MBS8506064)的浓度,通过酶标仪(MK3 LAB芬兰)检测450nm的吸光度,所有吸光度结果通过标准曲线标准化,计算出样品水平。
1.5肾小管细胞活力检测(MTT检测)取96孔板共培养实验中的肾小管上皮(约1×104)细胞,置37℃、5%CO2空气及100%湿度的细胞培养箱中培养24h。加入适当浓度的受试化合物并适当条件下培养24h。每孔加50μl1×MTT(KeyGEN KGA312),在37℃孵育4h。吸出上清液,每孔加150μl DMSO,酶标仪在570nm波长处检测每孔的光密度(OD值)。细胞的存活率:将各测试孔的OD值减去本底OD值(完全培养基加MTT,无细胞)或空白药物孔OD值(完全培养基加受试药物的不同稀释度加MTT,无细胞),各重复孔的OD值取均数±标准差(±s)。细胞的存活率以T/C%表示,T为加药细胞的OD值,C为对照细胞的OD值。细胞存活率%=(加药细胞OD/对照细胞OD)×100%。
1.6RT-qPCR检测AChE的mRNA相对表达用TRIzol液(Invitrogen,美国)提取肾内巨噬细胞总RNA,RNA的浓度和纯度用紫外分光光度计检测其在260和280nm的吸光度。逆转录反应用PrimeScript RT reagent Kit(TaKaRa Biotechnology-Co,Ltd,上海,中国)完成,每反应10μl总体系。RT-qPCR用SYBR Premix Ex TaqⅡ kit(TaKaRa BiotechnologyCo,Ltd,上海,中国)完成,每反应20μl体系。PCR扩增用一步法定量PCR系统完成(Applied Biosystems,Foster City,CA,美国),AChE mRNA的表达水平参照标准化的β-actin内参,数据采用2-ΔΔCt表示[8],并评估样本间差异。上述各试剂盒操作根据供应商操作说明。AChE和β-actin引物见表1。

表1 AChE和β-actin引物
1.7Western Blot分析为了检测AChE和对照GAPDH蛋白,用RIPA提取肾巨噬细胞总蛋白,取相同量的蛋白上样,电泳后,将一抗用TBST(SIGMA)稀释至1:500(在1.5ml离心管中);从封闭液中取出膜,室温下孵育1~2h后,用TBST在室温下脱色摇床上洗两次,每次10min;再用TBS洗一次,10min;用辣根过氧化物酶(HRP Abcam美国)标记的二抗稀释液(1:500)室温下孵育1h,用TBST在室温下脱色摇床上洗三次,每次10min,用ECL发光试剂盒检测。结果定量分析用Gel pro4.0版凝胶光密度分析软件进行分析,测其IOD(integrated optical density)累积光密度。
1.8统计学处理采用SPSS 18.0软件进行数据分析。数据以均数±标准差(±s)表示,两组间的比较采用独立样本t检验分析,P<0.05为差异具有统计学意义。
2 结果
2.1缺糖缺氧对肾内巨噬细胞培养上清液炎症介质及肾小管细胞活力的影响为了明确急性缺糖缺氧诱导的肾内炎症介质的表达,我们构建了缺糖缺氧肾内巨噬细胞模型,使用ELISA法检测细胞培养悬浮液中TNF-α,IL-1β和IL-10浓度。结果显示OGD组的TNF-α,IL-1β及IL-10表达均高于对照组(见图1A、B、C)。通过MTT法检测各实验组肾小管细胞的活力,结果发现与对照组(Normal cell)相比,10%XB+OGD组肾小管细胞活力明显降低(P<0.01)(见图1D)。

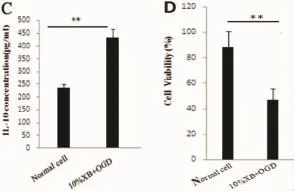
图1 急性缺糖缺氧对大鼠肾脏巨噬细胞炎症因子浓度及肾小管的活力的影响
2.3缺糖缺氧对肾巨噬细胞AChE mRNA及蛋白水平表达的影响为明确缺糖缺氧对胆碱能通路的影响,我们检测了两组乙酰胆碱酯酶mRNA及蛋白的表达。结果表明:肾巨噬细胞经过缺糖缺氧处理后,与对照组相比,AChE mRNA上调了(3.82± 0.73)倍,差异有统计学意义(P<0.01)(图2A),并且细胞内AChE蛋白水平也升高了(2.17±0.46)倍(图2B),AChE蛋白表达结果定量分析如 图2C所示。
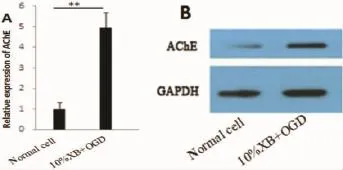
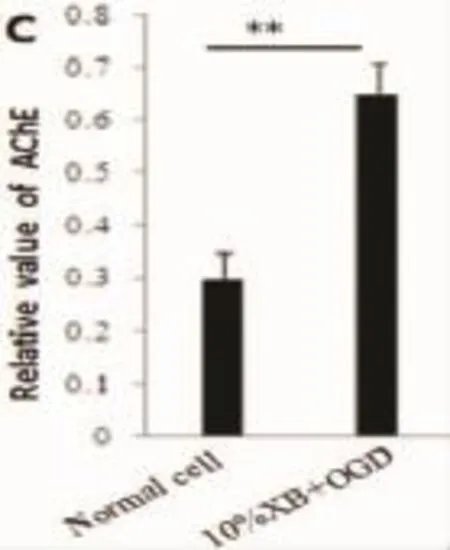
图2 急性缺糖缺氧对大鼠肾脏巨噬细胞AChE mRNA及蛋白的影响
3 讨论
2013年针对全国44家医疗机构的急性肾损伤流行病学调查显示有约25.3%患者的血清肌酐是正常值的2倍或以上,其中约7.0%患者怀疑有急性肾损伤,明显高于西方发达国家1.9%水平[9,10]。急性缺氧是急性肾损伤的重要原因。缺氧诱导的炎症细胞浸润在缺氧性肾损伤中起重要作用,其中巨噬细胞的肾脏浸润,显得尤为重要[11]。新近的研究发现,巨噬细胞在不同的微环境中有不同的表型,M1促进炎症反应,加重组织损伤,M2减轻炎症反应,促进组织修复等,巨噬细胞的极化状态对组织的损伤与修复起重要作用[12]。肾脏发生缺血性损伤时,循环中的炎症介质进入组织,改变了肾脏间质巨噬细胞的微环境。其中TNF-α、IL-1β可以促使巨噬细胞表现为M1型,促进炎症反应,IL-4、M-CSF、TGF-β可以促使巨噬细胞表达M2型,抑制炎症反应,促进组织修复[13]。也有研究显示iPSC-MSC来源的外泌体可抑制内毒素诱导的肺泡巨噬细胞分泌炎性因子,减轻肺损伤[14]。因此,探讨肾脏内的巨噬细胞的微环境对急性肾损伤的治疗至关重要。
迷走神经的胆碱能通路是一种及时的神经调节通路,激活此通路能有效减少炎性因子的合成和释放,对缺血再灌注损伤的全身和局部的炎症反应具有明显地抑制作用[15]。胆碱能神经末梢分泌乙酰胆碱,激活单核-巨噬细胞上的烟碱型乙酰胆碱受体7,抑制单核-巨噬细胞的活化,减少TNF-α等促炎因子的释放[16]。神经元细胞分泌的乙酰胆碱酯酶 (acetylcholinesterse,AChE)精准调控乙酰胆碱。避免神经末梢乙酰胆碱的集聚,对维持乙酰胆碱的动态稳定起积极作用。研究显示AChE不仅存在于神经元细胞中,也存在于免疫细胞上,其中巨噬细胞表达最丰富[7]。当机体受到不良刺激,组织的巨噬细胞能否调节AChE的分泌,适度调节局部的炎症反应尚不清楚。
为了明确肾脏微环境、炎症与胆碱能的关系,了解巨噬细胞在肾脏疾病的发展中的重要作用,我们构建OGD模型及肾脏巨噬细胞-上皮细胞共培养体系,以模拟急性缺血缺氧性所致肾小管损伤的病理过程。我们在肾脏巨噬细胞与小管上皮细胞共培养体系中,低糖低氧处理1h再正常培养至24h。结果发现:与对照组相比,OGD组细胞养上清液中TNF-α、IL-1β浓度水平明显增高,两者之间的差异有统计学意义(P<0.05),OGD组肾脏巨噬细胞中AChE mRNA和蛋白水平比正常对照组上调了(3.82±0.73)和(2.17±0.46)倍,并且肾小管细胞活力明显降低。说明缺糖缺氧促进巨噬细胞表面的胆碱酯酶的表达,通过调节炎症介质介导了缺糖缺氧性肾小管细胞损伤。
综上所述,本研究证实了缺糖缺氧增强了巨噬细胞分泌乙酰胆碱酯酶,间接减弱了胆碱能的抗炎作用,改变了肾脏间质细胞周围的微环境。我们推测,这种微环境的改变,使巨噬细胞表现为M1型,加剧了急性缺氧性炎症损伤。因此,我们将进一步探讨胆碱酯酶与巨噬细胞的表型之间的关系,为调控巨噬细胞的表型,减轻急性缺糖缺氧性肾损伤提供新的线索。
[1]Wang Y,Chang J,Yao B,et al.Proximal tubule-derived colony stimulating factor-1 mediates polarization of renal macrophages and dendritic cells,and recovery in acute kidney injury[J].Kidney Int,2015,88(6):1274-1282.
[2]Zuk A,Bonventre JV.Acute Kidney Injury[J].Annu Rev Med. 2016,67:293-307.
[3]Coca SG,Singanamala S,Parikh CR.Chronic kidney disease after acute kidney injury:a systematic review and meta-analysis[J].Kidney Int,2012,81(5):442-448.
[4]Cao Q,Harris DC,Wang Y.Macrophages in kidney injury,inflammation,and fibrosis[J].Physiology(Bethesda),2015,30(3):183-94.
[5]Tracey KJ.The inflammatory reflex[J].Nature,2002,420:853-859.
[6]Wang H,Yu M,Ochani M,et al.Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation[J].Nature,2003,421:384-388.
[7]Tracey KJ.Physiology and immunology of the cholinergic antiinflammatory pathway[J].J Clin Invest 2007,117:289-296.
[8]Livak J,Schmitten TD.Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T))Method[J].Methods,2001,25(4):402-408.
[9]Chen LX,Koyner JL.Biomarkers in Acute Kidney Injury[J].Crit Care Clin,2015,31(4):633-648.
[10]Yang L,Xing GL,Wang L,et al.Acute kidney injury in China:a cross-sectional survey[J].Lancet,2015,386(10002):1465-1471.
[11]Akcay A,Nguyen Q,Edelstein CL.Mediators of inflammation in Acute Kidney Injury[J].Mediators Inflamm,2009,2009:137072.
[12]Hume DA.The Many Alternative Faces of Macrophage Activation [J].Front Immunol,2015,6:370.
[13]Ricardo SD,van Goor H,Eddy AA.Macrophage diversity in renal injury and repair[J].J Clin Invest,2008,118(11):3522-3530.
[14]刘芬,李勇,彭菲菲,等.iPSC-MSC来源的外泌体对LPS刺激肺泡巨噬细胞产生炎性因子的影响.实验与检验医学,2016,34 (1):4-7.
[15]Inoue T,Abe C,Sung SJ,et al.Vagus nerve stimulation mediates protectionfromkidneyischemia-reperfusioninjurythrough α7nAChR+splenocytes[J].J Clin Invest,2016,126(5):1939-1952.
[16]Inoue T,Okusa MD.Neuroimmune Control of Acute Kidney Injury and Inflammation[J].Nephron,2015,131(2):97-101.
Acute oxygen and glucose deprivation promotes inflammatory injury of renal tubular cells by enhancing the expression of cholinesterase
WU Ming1,WU LeFeng1,LI Mingli2,LU Junfu1,LAI Kai1,XU Ji3,LIU Fen4,FENG Yongwen1.1.Department of Crit-ical Care Medicine of Shengzhen Second Hospital,Shenzhen 518035,China;2.Department of interventional therapy of Shengzhen Second Hospital,Shenzhen 518035,China;3.Department of Central laboratory of Shengzhen Second Hospital,Shenzhen 518035,China;4.the First Hospital Affiliated to Nan chang University,Nanchang 330006,China.
Objective To investigate the injury mechanism of renal tubular cells induced by acute oxygen and glucose deprivation.Methods Isolation and culture of rat kidney macrophages and renal epithelial cells,constructing co-cultivating model of lacking Oxygen and sugar(Oxygen and glucose deprivation,OGD),Cells were devided into control group and OGD group,and were given OGD treatment for 1 hour,and then carried out normal culture for up to 24 hours in each group.the expression of TNF alpha,IL-1 beta,IL-10 in supernatant fluid was detected by ELISA,the viability of renal tubular cells was determined by MTT,the expression of mRNA and protein of acetylcholine esterase(AChE)were determined by RT-qPCR and Western Blot respectively. Results The levels of TNF alpha(pg/ml)in the supernatant fluid in cultivation system were(231.67±36.28)in control group VS (428.67±43.16)(P<0.05)in OGD group,the levels of IL-1β(pg/ml)were(116.67±21.64)in control group VS(219.63±43.86)in OGD group(P<0.05),the levels of IL-10(pg/ml)were(235.67±39.35)in control group VS(432.67±49.72)in OGD group(P<0. 01).The viability of renal tubular cells was(88.41±18.25)VS(46.98±13.87)(P<0.01);The levels of mRNA and protein of AChE in OGD group were higher than those in control group,they were raised(3.82±0.73)and(2.17±0.46)times respectively(P<0.01). Conclusion Acute oxygen and glucose deprivation enhances the expression of cholinesterase in renal macrophages,the acute injury of renal tubular cells induced by OGD was mediated through inflammatory mediators.
Oxygen and glucose deprivation;Cholinesterase;Inflammatory mediators;Renal tubular cell viability
R692.6,R-332
A
1674-1129(2016)04-0412-04
10.3969/j.issn.1674-1129.2016.04.002
国家自然科学基金(81101410);广东省医学科学技术研究基金(A2016353);广东省深圳市科技创新委项目(JCYJ20130401112313541;JCYJ20150330102401099)深圳市第二人民医院基础-临床桥梁项目(2015)
吴明,男,1976年11月出生,医学硕士,副主任医师,主要从事于急性肾损伤的基础与临床研究。
冯永文,男,1959年12月出生,医学硕士,主任医师,硕士研究生导师,主要从事于危重患者的救治。E-mail:boshiyy@126.com
(2016-05-05;
2016-07-05)
猜你喜欢
中学生物学(2021年8期)2021-11-02
中国果业信息(2019年1期)2019-01-05
中成药(2018年7期)2018-08-04
中国畜牧兽医文摘(2018年6期)2018-07-28
哈尔滨医药(2016年3期)2016-12-01
中国卫生标准管理(2015年16期)2016-01-20
中外医疗(2015年11期)2016-01-04
科学启蒙(2015年8期)2015-08-07
医学研究杂志(2015年8期)2015-06-22
医学研究杂志(2015年12期)2015-06-10
