
基于3d过渡金属的氧电极材料研究进展
2016-08-23 09:19张慧娟
有色金属材料与工程 2016年3期
张 旭, 张慧娟
(上海理工大学 材料科学与工程学院, 上海 200093)
基于3d过渡金属的氧电极材料研究进展
张旭,张慧娟
(上海理工大学 材料科学与工程学院, 上海200093)
氧还原催化剂及缓慢的阴极氧还原动力学是制约低温燃料电池商业化的关键瓶颈因素之一.非贵金属氧还原催化剂是近年来低温燃料电池最受关注的研究热点之一.在简要介绍燃料电池及氧还原反应机理的基础上,详细地综述了近年来低温燃料电池用3d过渡金属基氧还原催化剂的主要研究进展,包括过渡金属大环化合物、过渡金属-氮/碳类化合物、过渡金属硫族化合物和过渡金属氧化物,总结了提高催化活性和稳定性、降低催化剂制备成本以及催化剂制备工艺等方面所取得的研究结果,并指出了各类催化剂目前尚待解决的问题和发展方向.
燃料电池; 氧还原反应; 氧电极; 催化剂; 3d过渡金属
燃料电池是一种不经过燃烧直接将燃料和氧化剂的化学能通过电化学反应方式转换成电能的发电装置,具有燃料多样化、环境友好、噪声低、可靠性高和维修方便等优点[1].在燃料电池工作系统中,氧气沿电极表面扩散进入电极内部,在催化剂的作用下发生还原反应.如何在提高氧电极活性的同时,降低氧还原反应的电化学极化及生产成本,寻求更高性价比的氧还原反应催化剂一直是燃料电池领域研究的重点之一[2].迄今为止,已报道的3d过渡金属基氧还原反应催化剂主要有过渡金属大环化合物、过渡金属-氮/碳类化合物、过渡金属硫族化合物和过渡金属氧化物,本文将主要介绍这些催化剂的近期研究进展.
1 氧还原反应机理

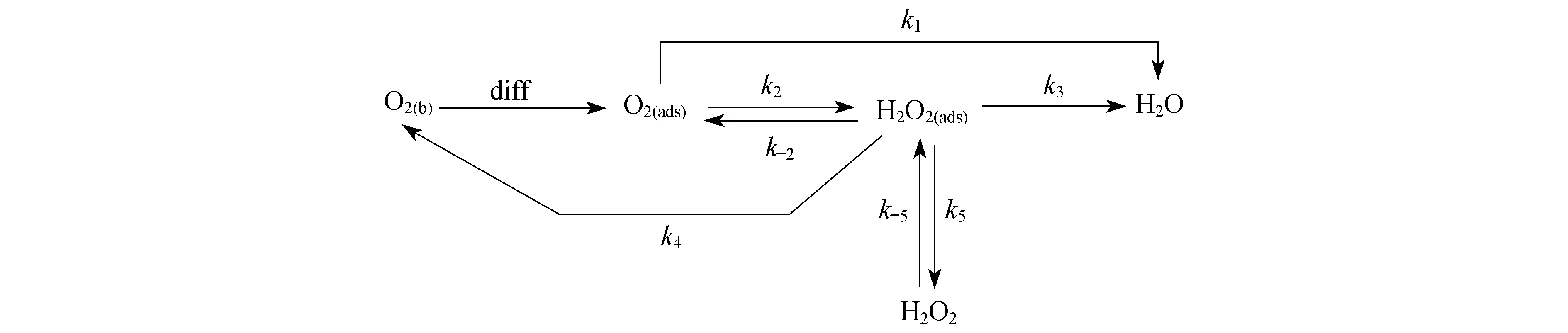
图1 电极上氧还原反应的Wroblowa过程图Fig.1 Wroblowa procedure chart of oxygen reduction reaction on electrode
2 氧电极材料研究现状
2.1过渡金属大环化合物
自1964年JASINSKI[5]发现酞菁钴具有催化氧还原活性后,其他一些金属大环化合物的氧还原催化作用被相继发现.研究表明,该类催化剂的性能主要取决于中心金属离子、大环配体、载体和热处理温度等因素.
2.1.1中心金属离子对催化性能的影响
中心金属离子对过渡金属大环化合物的氧还原催化活性起着决定性的作用[6-10].对过渡金属酞菁化合物来说,中心金属离子对氧还原催化活性的影响排序为Fe>Co>Ni>Cu≈Mn[6].CTÉ等[8]发现过渡金属氢氧化物对氧还原催化性能的影响是Cr>Fe>Co>>V.以过渡金属醋酸盐、氨气和四羧酸二酐为原料制备的催化剂对氧还原催化性能的影响是Fe>Co>Cr>Mn[10].YANG等[10]报道了TM-C-N对氧还原催化性能的影响:(1) 0.1M HClO4,Fe>Co>Cr>Ni>Mn>V;(2) 0.1M KOH,Co>Ni>Mn>V>Cr.
中心金属离子的用量也会影响催化剂的性能,而且使用不同金属前驱体将影响金属的最优载量.BRON等[11]发现含Fe质量分数为2%时铁邻菲罗啉催化剂的性能最好.LALANDE和FAUBERT等[12-13]研究发现CoPc中Co的最佳用量是质量分数为3.5%,而FeTPP中Fe的最佳用量是质量分数为4%.GUILLET等[14]研究发现CoTMPP/Vulcan XC72催化剂中Co的最佳用量是质量分数为0.2%.ZHANG等[15]研究了FeTPTZ中Fe用量对催化剂性能的影响,得到Fe的质量分数为4.7%的FeTPTZ催化性能最好.
2.1.2大环配体对催化性能的影响
对于给定的中心金属离子,氧还原催化活性在很大程度上取决于配体.对Fe而言,其活性为N>N2O2>N2S2>O4≈S4;但对Co来说,其活性为N2O2>N4>N2S2>O4≈S4[16].陶建中等[17]发现:与MTPP相比,MTNPP还原的E1/2均向正移.SONG等[18]发现在卟啉环的中心位置无取代基的钴卟啉具有更正的氧还原电位.GOUÉREC等[19]研究发现,随着大环上取代基电子接受能力的提高,催化剂的活性将降低.YU等[20]指出在卟啉环外围修饰了含Ru(Ⅱ)或Os(Ⅱ)的有机复合物的钴卟啉氧还原过程按4e反应进行的催化活性比修饰之前大大提高.BILOUL等[21]对不同的钴卟啉催化剂进行了100 mA/cm2恒电流工作300 h的PEMFC性能测试.
2.1.3载体对催化性能的影响
C载体的结构和表面物理化学性质与催化活性密切相关.WANG等[22]发现采用HNO3和NH3同时预处理的催化剂活性要比单独用HNO3或NH3预处理活性都高.SUBRAMANIAN等[23]认为用硝酸预处理的C载体表面引入了含氧官能团,这有利于提高钴乙二胺分子在C载体表面的分散度.C载体表面N含量的增加可以有效地提高催化剂的活性,而且N含量和C载体质量的减少是由于热处理过程中C载体与NH3反应,破坏了C载体的结构,并在其表面留下了N原子,C载体越无序,得到的催化剂活性越好[24].
C载体类型和负载方式对C载过渡金属大环化合物的氧还原催化性能有很大的影响.KUNDU等[25]研究得到C载体对邻菲罗啉催化性能的影响为:Black Pearls(1 500 m2/g)>Printex XE 2(900 m2/g)>Vulcan XC(254 m2/g).载体表面积和孔径分布不是决定催化剂活性的因素,催化剂活性与表面N含量直接相关,N含量越高,催化活性越好[26].含N条件下高能球磨石墨可以减小催化剂中晶体的颗粒尺寸,增加N含量、表面积和石墨的无序性,从而提高催化剂的活性[27].
2.1.4热处理对催化性能影响
高温热处理是改善大环化合物氧还原催化剂的催化活性和稳定性的有效途径.LEFVRE等[28]认为,最佳的催化活性发生在500~700 ℃,催化活性位是MN4/C;另外一个活性位为“高温活性位”,温度大于800 ℃.FAUBERT等[29]分别将FeTPP和CoTPP吸附在碳黑上,在Ar下进行热处理.当温度在500~700 ℃时,活性位为MN4或是一些分子碎片中包含了部分N与金属键合物,其稳定性较差;当温度大于900 ℃时,具有更好的活性和稳定性,且N与金属的键联消失,大量的金属颗粒被石墨包围.ALVES等[30]发现CoPc/C在850 ℃热处理后催化活性最高,此时存在直径为2 nm的Co簇.LALANDE等[31-32]把酞菁铁和四羧酸酞菁铁负载到碳黑上,并在氩气下100~1 100 ℃热处理,发现900 ℃以上的热解产物具有高催化活性和稳定性.GOJKOVIC等[33-35]对FeTMPP-Cl/BP研究表明:在200~400 ℃范围内开始分解;当T>400 ℃时,Fe(Ⅲ)/Fe(Ⅱ)氧化还原峰电对消失;当T>700 ℃时,有金属Fe粒子形成,此时循环伏安曲线上存在一个氧化峰,这是热处理形成的金属Fe的溶解过程.热处理温度为600~800 ℃时,钴卟啉具有最好的活性[36].酸性中,Co-C-N在临界热处理温度725 ℃处开始表现有催化活性,此时催化剂转变成β-Co和含N石墨碳的异相结构;碱性中,Co-C-N在500 ℃就开始表现有催化活性,在850 ℃处活性最佳[37].
2.1.5催化活性位探讨
催化活性位的确认一直是过渡金属大环化合物催化剂研究中倍受关注的课题.关于高温热解条件下的催化活性位存在很多争议[1,30,38-40]:(1) 高温热解后的催化活性位仍然是FeN4结构,如图2所示;(2) 高温热处理后残留的N与吸附在C表面上的过渡金属离子相互作用形成C-Nx-M物种,但FAUBERT认为Fe离子与C-Nx基团相结合对ORR的催化活性无影响;(3) N原子和过渡金属原子共同构成了催化活性位,而单纯由含N或过渡金属前驱体制备的物质无催化活性;(4) 热处理后形成具有化学表面基团的高活性C,并认为金属的存在只是催化了这种高活性C的形成,金属可能是起到在高温裂解含N和C前驱体时催化形成活性位的物质,更多的石墨边缘位和吡啶氮有利于提高催化活性,Fe颗粒的形成可能有利于边缘位C纳米结构的生长,如图3所示;(5) 在高温热解下形成的石墨包覆金属颗粒具有催化活性,石墨层可保护金属颗粒免于外界环境的腐蚀.
2.2过渡金属 - 氮/碳类化合物
研究表明,在高温热裂解下不使用大环化合物作前驱物也可以得到对氧有活性的催化剂.GUPTA等[41]首次报道了采用聚丙烯氰(PAN)、金属Co或铁盐和C载体混合后在惰性气氛下高温热处理制备氧还原催化剂的研究.该方法与常规的以含N4结构的大环化合物为前躯体的催化剂制备方法相比,不仅制备工艺简单,而且清洁无污染,因而引起了国内外学者的广泛兴趣.YE等[42]报道了通过碳化含有铁盐或钴盐的多孔PAN来制备Fe或Co气凝胶纳米复合物催化剂的方法.BRON等[43]报道了在Ar或NH3氛围中热处理碳载铁邻二氮杂菲化合物制备氧还原催化剂的研究.WEI等[44]通过裂解高比表面C、乙腈和硫酸钴的混合物来制备催化剂,结果表明,催化活性与N和Co的含量无关,但采用多步法可以得到性能较好的催化剂.OKADA等[45]报道了N、N′-二-8-喹啉三甲基二胺钴和N、N′-二-8-喹啉苯基二胺钴的催化性能随着热处理温度的升高而增加.ALVES等[46]以聚甲基乙烯基酮(PVK)、聚丙烯酸(PAA)和聚苯乙烯(PE)为配体考察了配位作用对催化剂性能的影响.BOUWKAMP-WIJNOLTZ等[47]提出了用乙酸钴、碳黑和各种含N前驱体制备氧还原催化剂的方法.EXAFS表明,这类催化剂有活性位CoN4,但同时也有金属Co的存在.JIANG等[48]研究发现ORR反应路径与配体的结构有关:2、4、6 -三甲基苯基配体是2e反应路径,2、6 -二异丙基苯基配体是4e反应路径.
另外,SIRK等[49]基于乙二胺和1、2 -苯乙二胺合成了两种Co基氧还原催化剂.YUASA等[50]用电化学沉积法在C颗粒表面修饰一层聚吡咯膜(PPY/C),并在其表面吸附Co离子,700 ℃热处理后获得了在酸性条件下稳定的Co-PPY/C.2006年,BASHYAM等[51]在《Nature》上报道了用化学还原法合成了Co-PPY-C作为PEMFC阴极催化剂的工作,如图4所示.

图2 MN4催化活性位示意图Fig.2 Schematic illustration of the MN4 catalytic active site
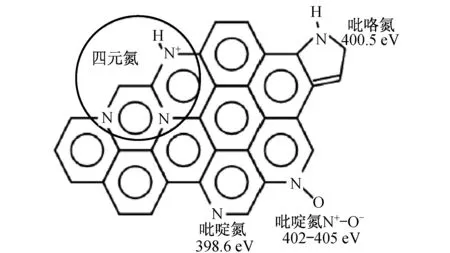
图3 不同类型N在C载体表面的示意图Fig.3 Schematic illustration of different types of N on the surface of carbon surport

图4 Co-PPY-C催化剂的结构示意图Fig.4 Structure illustration of the Co-PPY-C catalyst
2.3过渡金属硫族化合物
过渡金属硫族化合物是由纳米过渡金属与硫族元素形成的簇合物,可以分为Chevrel相结构和无定型相结构两种.其中,Chevrel相结构的过渡金属硫族化合物具有一个中心正八面体的金属簇,周围环绕着8个组成立方形的硫族原子,其通式表达为Mo6X8或Mo6-xMxX8(X=S、Se、Te、SeO;M=Os、Re、Rh、Ru)和MxMo6X8,具有很好的导电性,通常是在高温高压中反应并经热处理等一系列操作得到的.无定型相的过渡金属硫族化合物是由2个相互独立的稳定相组成,通常的制备方法是以羰基金属M(M=Ru、W、Mo、Re)和硫族元素X(X=S、Se、Te)为原料,在有机溶剂中边回流边反应得到.这种制备方法与Chevrel相簇合物的制法相比,毒性小,操作简便.过渡金属硫族化合物的催化活性主要受簇合物中过渡金属的化学计量比影响,如Mo3.7Ru2.3Se8>Ni0.85Mo6Te8>Mo4.2Ru1.8Se8.簇合物中掺杂的客体金属使簇合物的弱态d态电子增加,不仅提高了簇合物催化氧还原的活性,而且提高了稳定性;但客体金属在催化反应中会部分释放出来形成氧化物,产生混合电位.
该类催化剂的氧还原机理的研究表明[52-54]:(1) 簇合物中过渡金属的协同作用决定催化活性,如Ru取代Mo得到的Mo4.2Ru1.8Se8对氧还原的催化活性大大优于非取代的Mo6Se8;(2) 簇合物的费米能带周围有高密度的弱态d电子,这对氧还原催化活性起主要作用;(3) 簇合物内原子间键距有利于氧的键合,并能在簇内原子间形成桥式结构,如Mo4.2Ru1.8Se8中同一原子簇的原子间最小键距d=0.271 nm,有利于氧的键合以及在簇内原子间形成桥式结构;(4)在氧与簇合物间的电子转移过程中,该类簇合物能够改变自身体积和成键距离,有利于氧的4e还原,96%以上的直接产物为水.进一步的研究表明:第一步的反应产物不是O2-而是还原氧与过渡金属簇合物的化学键合,过渡金属间的键距d增大,促进O—O键断裂,释放出OH-.该机理是以簇合物中价电子的标准数和簇合物中金属 - 金属键距的关系为基础.
2.4过渡金属氧化物
过渡金属氧化物主要分为钙钛矿型和尖晶石型,其中钙钛矿型氧化物的研究较多.
2.4.1钙钦矿型
钙钛矿型氧化物结构通式为ABO3(A=La、Ca、Sr、Ba;B=Co、Fe、Mn、Ni、Cr),理想的ABO3钙钛矿结构是立方的.离子半径大的稀土金属离子占据A位置,周围有12个氧离子配位,A与O形成最密堆积,A位的稀土元素很少直接作为活性点起催化作用,大多数只是作为晶体稳定点阵的组成部分而间接发挥作用;B位为过渡金属元素,B离子周围有6个氧离子,催化活性主要取决于该位.在氧化物骨架中,A和B的半径分别为rA>0.090 nm,rB>0.051 nm.对于某种B离子,A离子的半径由下式限制[1,54]:
0.75<(rA+rB)/2(rB+rO)<1.00
(1)
式中:rA、rB分别为A、B位离子半径;rO为氧离子半径.
纯粹的ABO3结构的催化活性并不高,可通过A位或B位金属的部分替换对组分原子价态进行控制.对于组分A,当A为La和Pr时,其催化活性最高,并且当A部分被Ca、Sr和Ba取代时,此类氧化物具有更好的电催化活性,稳定性也得到很大的提高[2].对于组分B,此类氧化物在碱性介质中对ORR催化活性的顺序为Co>Mn>Fe,稳定性的顺序为Fe>Mn>Co,所以当B为Mn时,氧化物有较好的活性和化学稳定性[2].LIU等[55]采用sol - gel法制备了Pr1-xCaxCrO3,发现Pr1-xCaxCrO3为单一的钙铁矿相,部分Ca2+取代Pr3+后,为了维持电荷平衡,Cr在晶体中呈混合价态,由Cr3+上升为Cr6+,增加了导电性,其中以Pr0.6Ca0.4CrO3的导电性最高.唐致远等[56]从LaNiO3的B位掺杂出发,采用溶胶-凝胶法制备了一系列LaNi1-yCoyO3型电催化剂,发现B位掺杂可明显提高催化剂B表面离子的浓度,提高电极电催化性能.顾军等[57]用溶胶-凝胶法制备了La1-xCaxFe1-yCoyO3,得到兼具高活性和稳定性的双功能氧电极,研究发现x=0.4时电催化活性最好.
钙钛矿型氧化物的合成方法主要有盐分解法、共沉淀法、氧化物混合热处理法和无定形羟酸前驱体法[1],这几种方法各有利弊,既要得到好的催化效果又要兼顾大规模工业生产的实用性,可根据实际需要选取不同的制备方法.WU等[58]对La0.6Ca0.4CoO3-x用真空退火处理,然后在空气中从800 ℃到100 ℃连续降温氧化处理,系统研究了氧化温度与催化活性的关系,发现Co的价态对退火条件很敏感.退火温度从800 ℃下降到300 ℃时,催化活性单调下降,氧的含量增加;继续下降时催化活性快速上升,ORR在200 ℃时达到最大值.SHIMIZU等[59]测试了用无定型柠檬酸前驱体法(ACP)及醋酸盐分解法(AD)制备La1-xCaxCoO3电极的电极性能,发现ACP法要优于AD法,因为用ACP法制成的La1-xCaxCoO3的表面积是用AD法制成的5~9倍,而且电极以100 mA/cm2充放电100个循环后,性能稳定.
2.4.2尖晶石型
分子式通式为AB2O4的尖晶石型过渡金属氧化物是另一类很有前景的催化剂.在这类化合物中,离子半径接近的过渡金属分别占据四面体和八面体空位,与氧配位.尖晶石过渡金属氧化物的催化活性来自与八面体配位的变价过渡金属离子氧化还原对[52-54].PIOS和PONCE等[60-61]对Cu-Mn、Mn-Co和Ni-Co尖晶石材料及其部分掺杂材料进行了深入研究,发现尖晶石型结构的催化活性取决于B位变价离子数的比例和与制备方式紧密相关的表面粗糙程度.HELLER-LING等[62]用双管道流电池电极对NixCo3-xO4氧还原催化活性进行的研究表明,当x=0或0.6
3 结 论
催化剂是氧电极的关键因素,制备性能优良、工作稳定的催化剂仍是研究者追求的目标.综上所述,经过国内外研究者的共同努力,燃料电池基于3d过渡金属的氧电极催化剂的种类繁多,对氧的还原都有催化活性,但真正投入实际使用的却很少.寻找廉价、高效的催化剂已成为提高氧电极性能的关键,这个问题一旦解决,必将大大推动燃料电池的发展.
[1]张慧娟.新型非贵金属螯合物氧还原催化剂制备及其电化学性能研究[D].上海:上海交通大学,2010.
[2]黄庆华,李振亚,王为.电池用氧电极催化剂的研究现状[J].电源技术,2003,27(z1):241-244.
[3]WROBLOWA H S,PAN Y C,RAZUMNEY G.Electroreduction of oxygen:a new mechanistic criterion[J].Journal of Electroanalytical Chemistry and Interfacial Electro-chemistry,1976,69(2):195-201.
[4]YEAGER E.Electrocatalysts for O2reduction[J].Electrochimica Acta,1984,29(11):1527-1537.
[5]JASINSKI R.A new fuel cell cathode catalyst[J].Nature,1964,201(4925):1212-1213.
[6]BECK F,DAMMERT W,HEISS J,etal.Electrocatalysis of the oxygen cathode by metal-phthalocyanines and-dibenzotetraazaannulenes[J].Zeitschrift für Naturforschung A:Journal of Physical Sciences,1973,28(6):1009-1021.
[7]van VEEN J A R,VISSER C.Oxygen reduction on monomeric transition metal phthalocyanines in acid electrolyte[J].Electrochimica Acta,1979,24(9):921-928.
[10]YANG R Z,STEVENS K,DAHN J R.Investigation of activity of sputtered transition-metal(TM)-C-N(TM=V,Cr,Mn,Co,Ni)catalysts for oxygen reduction reaction[J].Journal of the Electrochemical Society,2008,155(1):B79-B91.
[11]BRON M,FIECHTER S,HILGENDORFF M,etal.Catalysts for oxygen reduction from heat-treated carbon-supported iron phenantrolinecomplexes[J].Journal of Applied Electrochemistry,2002,32(2):211-216.
[12]LALANDE G,TAMIZHMANI G,CTÉ R,etal.Influence of loading on the activity and stability of heat-treated carbon-supported cobalt phthalocyanineelectrocatalysts in solid polymer electrolyte fuel cells[J].Journal of the Electrochemical Society,1995,142(4):1162-1168.
[13]FAUBERT G,CTÉ R,GUAY D,etal.Iron catalysts prepared by high-temperature pyrolysis of tetraphenylporphyrins adsorbed on carbon black for oxygen reduction in polymer electrolyte fuel cells[J].Electrochimica Acta,1998,43(3-4):341-353.
[14]GUILLET N,ROUÉ L,MARCOTTE S,etal.Electrogeneration of hydrogen peroxide in acid medium using pyrolyzed cobalt-based catalysts:influence of the cobalt content on the electrode performance[J].Journal of Applied Electrochemistry,2006,36(8):863-870.
[15]ZHANG L,LEE K,BEZERRA C W B,etal.Fe loading of a carbon-supported Fe-N electrocatalyst and its effect on theoxygen reduction reaction[J].Electrochimica Acta,2009,54(26):6631-6636.
[16]VASUDEVAN P,SANTOSH,MANN N,etal.Transition metal complexes of porphyrins and phthalocyanines as electrocatalysts for dioxygen reduction[J].Transition Metal Chemistry,1990,15(2):81-90.
[17]陶建中,王三虎,娄天军,等.四(对-硝基)苯基卟啉合钴、锰、铁、锌配合物电化学和光谱电化学性质的研究[J].化学研究与应用,1999,11(3):268-271.
[18]SONG E,SHI C N,ANSON F C.Comparison of the behavior of several cobalt porphyrins as electrocatalysts for the reduction of O2at graphite electrodes[J].Langmuir,1998,14(15):4315-4321.
[19]GOUÉREC P,BILOU A,CONTAMIN O,etal.Dioxygen reduction electrocatalysis in acidic media:effect of peripheral ligand substitution on cobalt tetraphenylporphyrin[J].Journal of Electroanalytical Chemistry,1995,398(1-2):67-75.
[20]YU H Z,BASKIN J S,STEIGER B,etal.Femtosecond dynamics and electrocatalysis of the reduction of O2:tetraruthenated cobalt porphyrins[J].Journal of the American Chemical Society,1999,121(2):484-485.
[21]BILOUL A,GOUÉREC P,SAVY M,etal.Oxygen electrocatalysis under fuel cell conditions:behaviour of cobalt porphyrins and tetraazaannuleneanalogues[J].Journal of Applied Electrochemistry,1996,26(11):1139-1146.
[23]SUBRAMANIAN N P,KUMARAGURU S P,COLON-MERCADO H,etal.Studies on Co-based catalysts supported on modified carbon substrates for PEMFC cathodes[J].Journal of Power Sources,2006,157(1):56-63.
[24]JAOUEN F,CHARRETEUR F,DODELET J P.Fe-based catalysts for oxygen reduction in PEMFCs-importance of the disordered phase of the carbon support[J].Journal of the Electrochemical Society,2006,153(4):A689-A698.
[25]KUNDU S,NAGAIAH T C,XIA W,etal.Electrocatalytic activity and stability of nitrogen-containing carbon nanotubes in the oxygen reduction reaction[J].The Journal of Physical Chemistry C,2009,113(32):14302-14310.
[26]JAOUEN F,MARCOTTE S,DODELET J P,etal.Oxygen reduction catalysts for polymer electrolyte fuel cells from the pyrolysis of iron acetate adsorbed on various carbon supports[J].The Journal of Physical Chemistry B,2003,107(6):1376-1386.
[27]PROIETTI E,RUGGERI S,DODELET J P.Fe-based electrocatalysts for oxygen reduction in PEMFCs using ballmilled graphite powder as a carbon support[J].Journal of the Electrochemical Society,2008,155(4):B340-B348.
[29]FAUBERT G,LALANDE G,CTÉ R,etal.Heat-treated iron and cobalt tetraphenylporphyrins adsorbed on carbon black:physical characterization and catalytic properties of these materials for the reduction of oxygen in polymer electrolyte fuel cells[J].Electrochimica Acta,1996,41(10):1689-1701.
[30]ALVES M C M,DODELET J P,GUAY D,etal.Origin of the electrocatalytic properties for O2reduction of some heat-treated polyacrylonitrile and phthalocyanine cobalt compounds adsorbed on carbon black as probed by electrochemistry and X-ray absorption spectroscopy[J].The Journal of Physical Chemistry,1992,96(26):10898-10905.
[31]LALANDE G,FAUBERT G,CTÉ R,etal.Catalytic activity and stability of heat-treated iron phthalocyanines for the electroreduction of oxygen in polymer electrolyte fuel cells[J].Journal of Power Sources,1996,61(1/2):227-237.
[32]LALANDE G,CTÉ R,TAMIZHMANI G,etal.Physical,chemical and electrochemical characterization of heat-treatedtetracarboxylic cobalt phthalocyanineadsorbed on carbon black as electrocatalyst for oxygen reduction in polymer electrolyte fuel cells[J].Electrochimica Acta,1995,40(16):2635-2646.
[36]OKADA T,GOKITA M,YUASA M,etal.Oxygen reduction characteristics of heat-treated catalysts based on cobalt-porphyrin ion complexes[J].Journal of the Electrochemical Society,1998,145(3):815-822.
[37]YANG R Z,STEVENS K,BONAKDARPOUR A,etal.Dependence of the activity of sputtered Co-C-N oxygen reduction electrocatalysts on heat-treatment temperature[J].Journal of the Electrochemical Society,2007,154(9):B893-B901.
[38]BOUWKAMP-WIJNOLTZ A L,VISSCHER W,van VEEN J A R,etal.On active-site heterogeneity in pyrolyzed carbon-supported iron porphyrin catalysts for the electrochemical reduction of oxygen:an in situ Mössbauerstudy[J].The Journal of Physical Chemistry B,2002,106(50):12993-13001.
[39]MATTER P H,OZKAN U S.Non-metal catalysts for dioxygen reduction in an acidic electrolyte[J].Catalysis Letters,2006,109(3-4):115-123.
[40]MATTER P H,ZHANG L,OZKAN U S.The role of nanostructure in nitrogen-containing carbon catalysts for the oxygen reduction reaction[J].Journal of Catalysis,2006,239(1):83-96.
[41]GUPTA S,TRYK D,BAE I,etal.Heat-treated polyacrylonitrile-based catalysts for oxygen electroreduction[J].Journal of Applied Electrochemistry,1989,19(1):19-27.
[42]YE S Y,VIJH A K.Non-noble metal-carbonized aerogel composites aselectrocatalysts for the oxygen reduction reaction[J].Electrochemistry Communications,2003,5(3):272-275.
[43]BRON M,RADNIK J,FIEBER-ERDMANN M,etal.EXAFS,XPS and electrochemical studies on oxygen reduction catalysts obtained by heat treatment of iron phenanthroline complexes supported on high surface area carbon black[J].Journal of Electroanalytical Chemistry,2002,535(1-2):113-119.
[44]WEI G,WAINRIGHT J S,SAVINELL R F.Catalytic activity for oxygen reduction reaction of catalysts consisting of carbon,nitrogen and cobalt[J].Journal of New Materials for Electrochemical Systems,2000,3(2):121-129.
[45]OKADA T,YOSHIDA M,HIROSE T,etal.Oxygen reduction characteristics of graphite electrodes modified with cobalt di-quinolyldiaminederivatives[J].Electrochimica Acta,2000,45(27):4419-4429.
[46]ALVES M C M,TOURILLON G.Influence of complexation processes on the catalytic properties of some polymer-based cobalt compounds for oxygen electroreduction[J].The Journal of Physical Chemistry,1996,100(18):7566-7572.
[47]BOUWKAMP-WIJNOLTZ A L,VISSCHER W,van VEEN J A R,etal.Electrochemical reduction of oxygen:an alternative method to prepare active CoN4catalysts[J].Electrochimica Acta,1999,45(3):379-386.
[48]JIANG J H,KUCERNAK A.Novel electrocatalyst for the oxygen reduction reaction in acidic media using electrochemically activated iron 2,6-bis(imino)-pyridyl complexes[J].Electrochimica Acta,2002,47(12):1967-1973.
[49]SIRK A H C,CAMPBELL S A,BIRSS V I.Oxygen reduction by sol derived[Co,N,C,O]-based catalysts for use in proton exchange membrane fuel cells[J].Electrochemical and Solid-State Letters,2005,8(2):A104-A107.
[50]YUASA M,YAMAGUCHI A,ITSUKI H,etal.Modifying carbon particles with polypyrrole for adsorption of cobalt ions as electrocatatytic site for oxygen reduction[J].Chemistry of Materials,2005,17(17):4278-4281.
[51]BASHYAM R,ZELENAY P.A class of non-precious metal composite catalysts for fuel cells[J].Nature,2006,443(7107):63-66.
[52]李旭光,邢巍,唐亚文,等.直接甲醇燃料电池阴极电催化剂的研究进展[J].化学通报,2003,66(8):521-527.
[53]刘洁,刘世斌,张忠林,等.DMFC用阴极抗甲醇催化剂[J].电池,2006,36(1):77-79.
[54]唐致远,宋世栋,刘建华.钙钛矿型双功能氧电极催化剂的研究进展[J].电源技术,2003,27(z1):233-237.
[55]LIU X M,SU W H,LU Z.Study on synthesis of Pr1-xCaxCrO3and their electrical properties[J].Materials Chemistry and Physics,2003,82(2):327-330.
[56]唐致远,宋世栋,刘建华,等.La1-xSrxNi1-yCoyO3双功能氧电极的电化学性能[J].物理化学学报,2003,19(9):785-790.
[57]顾军,隋升,李光强,等.La1-xCaxFe1-yCoyO3对氧气还原的催化活性[J].无机材料学报,1999,14(4):618-623.
[58]WU N L,LIU W R,SU S J.Effect of oxygenation on electrocatalysis of La0.6Ca0.4CoO3-xin bifunctional air electrode[J].Electrochimica Acta,2003,48(11):1567-1571.
[59]SHIMIZU Y,UEMURA K,MATSUDA H,etal.Bi-functional oxygen electrode using large surface area La1-xCaxCoO3for rechargeable metal-air battery[J].Journal of the Electrochemical Society,1990,137(11):3430-3433.
[60]RIOS E,GAUTIER J L,POILLERAT G,etal.Mixed valency spinel oxides of transition metals and electrocatalysis:case of the MnxCo3-xO4system[J].Electrochimica Acta,1998,44(8-9):1491-1497.
[62]HELLER-LING N,PRESTAT M,GAUTIER J L,etal.Oxygen electroreduction mechanism at thin NixCo3-xO4spinel films in a double channel electrode flow cell(DCEFC)[J].Electrochimica Acta,1997,42(2):197-202.
[63]NGUYEN-CONG H,de la GARZA GUADARRAMA V,GAUTIER J L,etal.Oxygen reduction on NixCo3-xO4spinel particles/polypyrrole composite electrodes:hydrogen peroxide formation[J].Electrochimica Acta,2003,48(17):2389-2395.
Research on Catalysts for Oxygen Electrode Based on 3d Transition Metal
ZHANG Xu,ZHANG Huijuan
(School of Materials Science and Engineering, University of Shanghai for Science and Technology, Shanghai 200093, China)
The electrocatalyst for oxygen reduction reaction(ORR) and the slow cathodic oxygen reduction reaction are the key bottleneck for the commercialization of low temperature fuel cell.In recent years,non-precious metal catalysts for oxygen reduction have gained particular interest in the research on low temperature fuel cells.In this paper,research progress on 3d transition metal-based electrocatalysts,including transition-metal macrocyclic compound,transition metal-nitrogen/carbon compound and transitional-metal chalcogenide and transitional-metal oxide for the oxygen electrode have been systematically reviewed in addition to a brief introduction of the reaction mechanism of ORR at cathode electrode and FCs.Research findings on how to improve the activity and durability of the catalysts,reduce costs of preparing catalysts,and to develop novel non-precious metal catalystsare were summarized.Problems were to be solved and the future of electrocatalyst were pointed out as well.
fuel cells; oxygen reduction reaction; oxygen electrode; catalysts; 3d transition metal
1005-2046(2016)03-0110-08
DOI:10.13258/j.cnki.nmme.2016.03.009
2015-04-27
国家自然科学基金资助项目(21406139);上海市自然基金项目(13ZR1429000);沪江基金(B14006)
张旭(1994—),男,硕士研究生. 主要从事碳基氧电极材料的研究. E-mail: 2804128773@qq.com
张慧娟(1980—),女,高级实验师. 主要从事电催化材料方面的研究. E-mail: hjzhang@usst.edu.cn
TM 911.46
A
猜你喜欢
热处理技术与装备(2022年1期)2022-11-29
金属热处理(2022年2期)2022-11-16
金属热处理(2022年1期)2022-03-15
军民两用技术与产品(2021年8期)2021-11-24
安徽工业大学学报(自然科学版)(2021年3期)2021-09-08
装备维修技术(2020年5期)2020-11-20
分析化学(2018年1期)2018-01-18
分析化学(2017年9期)2017-10-16
中学化学(2015年9期)2016-04-14
中学化学(2015年6期)2015-06-18
