
12个亨廷顿病家系的临床特点及家系分析
2016-08-03 02:14苏凤娟曾译萱裴中梁秀龄李洵桦JeanMarcBurgunder
中国神经精神疾病杂志 2016年1期
苏凤娟曾译萱△裴中△梁秀龄△李洵桦△ Jean-Marc Burgunder
12个亨廷顿病家系的临床特点及家系分析
苏凤娟*曾译萱*△裴中*△梁秀龄*△李洵桦*△Jean-Marc Burgunder◎
目的探讨亨廷顿病的临床症状、突变基因以及家系中遗传规律等。方法 收集2013-2014年至中山大学附属第一医院就诊的亨廷顿病患者,绘制完整的家谱图并记录详细的临床资料。对每位患者予IT15基因诊断。对其症状改变,UHDRS评分、行为学进行评估。结果 2013年1月至2014年12月共收治12个亨廷顿病家庭,12例先证者中有2例处于症状前状态,1例青少年亨廷顿病患儿。所有确诊患者的IT15突变基因的CAG重复序列大多在40~60之间,已发病患者的起病年龄13~54岁不等。其家系分析中发现,父系遗传有遗传早现现象。12例家系中,发病晚期的患者有较典型的舞蹈症状、智能减退、精神症状的临床表现,早期发病患者临床症状多变,或以情绪与智能损害为首发症状。结论 晚期亨廷顿病患者临床表现较典型,但早期亨廷顿病的症状多样且不典型,临床诊断困难,行为学改变,影像学检查有重要的参考价值,基因诊断是该疾病确诊的重要方式。
亨廷顿病基因诊断影像学行为问题
【Abstract】Objective To investigate the clinical manifestation,inherited pattern and the related factor of Huntington disease families.Method The clinical data from 12 HD families was collected from 2013-2014.Patients received the genetic test and neurological evaluation including motor,cognitive and problem of behavior.Results There were 12 patients having the IT15 gene dynamic mutations,including 1 Juvenile Huntington disease patient and 3 pre-symptomatic mutant gene carriers.The average CAG repeats of these patients was between the range of 40 to 60,and the average onset age ranged from 13 to 54 year-old.Positive family history and genetic anticipation could be observed.Patients presented with different clinical manifestations at the early stage while had typical chorea movements,declined cognitive and psychiatric symptoms at the late stage of the illness.Conclusions There are typical triad symptoms in the late stage but not in the early stage nor pre-symptom stage illness.Clinical manifestation and the neuroimaging are both of great reference value,and the genetic test is essential for final diagnosis.
【Key Words】Huntington disease Genetic testNeuroimaging Behavior problem
亨廷顿病(Huntington disease,HD)是常染色体显性遗传性疾病。最早于1872年由美国医生George Huntington报道。HD是一种基底核和大脑皮质的变性疾病,其病变基因位于4p16.3中IT15基因上[1]的一段多态性的三核苷酸(又称三联体triplet)CAG的异常重复序列:CAG拷贝数在正常人群中为11~34,平均为19,但在患者中异常增多可达42~100个。临床症状上,HD患者多在成年后发病,有特征性的舞蹈样症状[2-3],即突然发生的、过度的、自发的并且持续存在的舞蹈样动作以及肌张力障碍等运动障碍,症状慢性进行性加重。通常还会伴有不同程度的精神异常和智能异常等表现。另外,约有3%~10%的患者在青少年期(通常在20岁之前)发病[4],主要表现为肌张力障碍-运动迟缓、癫痫、智能下降等,其突变基因的CAG扩增多在60次以上,称为青少年型亨廷顿病(Juvenile Huntington Disease,JHD)。本研究主要对成年型HD患者的临床症状(运动及非运动)进行分析,总结中国HD患者临床症状特点,为HD早期临床诊断提供相关的依据。
1 资料与方法
1.1一般资料 收集2013~2014年收治的有症状的亨廷顿病患者21例(包括基因诊断者以及临床诊断者),来自12个家系,均为汉族;男9例,女12例;其中12例行基因检测确诊,9例为临床诊断(均有阳性家族史)。除此之外,另有基因诊断明确的症状前患者3例。
1.2诊断标准 根据中华医学会神经病学会帕金森病及运动障碍学组亨廷顿病的诊断指南(2011年)[5],对其一级家属有阳性家族史和特征性舞蹈样运动症状、认知和精神症状,伴或不伴有家族中有基因确诊的HD者,可做出临床诊断,也可进一步进行基因诊断;如无阳性家族史,或临床症状不典型,结合其影像学检查等可通过基因检测以进一步确诊。
2 结果
2.1一般情况 本研究收录的12个HD家系中,有21例已发病的HD患者以及3例基因诊断的未发病患者,已发病患者中有12例获得基因确诊,另9例根据其阳性家族史以及临床症状予临床诊断。已发病患者中除1例青少年发病者,均为成年发病,起病年龄为29~54岁不等,平均病程为5年。
除JHD患儿以智能下降就诊外,所有成年发病患者均以肢体或者躯体的舞蹈样症状为初次就诊原因。对于有明显运动症状的患者,追问病史时有6/10的先证者情绪、性格的改变早于运动症状出现,并且均被家人忽略。其中2例在起病早期有智能损害。
12个家系先证者情况统计如下(见表1):其中有4个无阳性家族史,其中3个(家系1、8、10)是由于家族信息追访不全,1例为JHD患儿(家系7),其父亲尚无运动症状以致病史采集中获得假阴性家族史。对已有基因诊断患者的IT15基因中CAG重复次数进行分析,发现其突变长链的CAG重复次数为多在40~60之间(除外青少年型HD患者);所有先证者发病年龄与CAG重复次数相关性分析中可发现,发病年龄与其长链CAG重复次数有负性相关趋势(图1,A)。
2.2部分HD家系家谱图分析 在有HD家族史的家系中,均可发现较典型的“遗传早现”现象。以家系3为例:Ⅰ1 50多岁发病,舞蹈样动作症状起病,具体发病时间以及死亡时间等不详;Ⅱ2于35岁发病,舞蹈样动作症状起病,去世前舞蹈症状严重影响生活;Ⅲ2、3、6、7均在在30岁左右发病,其中Ⅲ3于发现有运动症状后4年去世;先证者Ⅳ4 于27岁起病,以情绪改变(易激惹)以及全身舞蹈样症状发病,现发病6年余,近来存在被害妄想。
家系5的先证者Ⅲ5(15岁)是一例基因确诊的JHD患者,初次就诊时诉家族中无HD家族史,家族中Ⅰ1在70多岁死亡且不伴有舞蹈症状。由于患者颅脑磁共振检查结果示有明显的尾状核头部的萎缩及侧脑室的扩大,予行HD基因检查,最终获得基因确诊(CAG重复次数为17/75)。其后对其父母进行HD基因检测发现其父亲Ⅱ5是完全外显突变者(CAG重复次数为18/41),但仍处于发病前状态。
2.3问题行为学改变 行为学改变同样是亨廷顿舞蹈症患者常见的症状之一,并且会随着病程进展而加重。取相应的11个行为学相关指标,并对所有患者进行评估:抑郁情绪、自杀观念、焦虑、易激惹、发怒或攻击行为、淡漠、刻板思维或行为、强迫障碍、妄想、幻觉以及定向力障碍。每个评定项目由严重性(相应症状的严重程度)以及频率(以症状于每周或者每个月出现的次数分级)综合表示。严重性以及频率均从1~4进行评估(严重性分级:0=无,1=可疑,2=轻度,但不造成问题,3=中度,症状造成问题,4=严重,看护着几乎无法忍受;频率分级:0=从不,1=较少,少于1周1次,2=有时,每周可达4次,3=经常,大多数时间,4=几乎每天且全天)。而相应的症状评估则以(严重性×频率)表示其最后的结果。得分越高,表明对应的症状越严重。

表1 15个家系先证者患者资料

图1 IT15基因突变长CAG重复次数与起病年龄的相关性分析以及部分基因分析结果显示 (A)所有先证者发病年龄与CAG重复次数的相关性分析,已发病患者的发病年龄与其长链CAG重复次数有负性相关趋势(R2=0.61)
对12个家系中已发病先证者的询问中发现有6/10在舞蹈样症状开始前有行为学的改变。3例已检测基因的未有运动症状的突变基因携带者的量表评估示有2例已有行为学改变。在所有已发病的家属中,有半数以上有运动症状患者已经出现问题行为学的改变。进一步对其中7例患者使用行为学评估量表PBA-s进行的评估,按照文献对其运动症状的严重程度予临床分期[7](症状前状态:没有运动症状出现;早期:有轻微运动症状,但大部分功能区仍活跃,依然可以工作或驾驶;中期:患者无法进行复杂的事件如独立工作;晚期:患者无法独自进行日常活动):患者1处于症状前状态,表现出易激惹以及发怒行为,自己尚能控制情绪,偶发淡漠;患者2、3、4在发病早期,有轻微舞蹈样症状,生活能自理,病史<5年,主要表现为明显易激惹以及偶发发怒、攻击行为,有被害妄想存在,1例有刻板行为;患者5、6、7处于HD发病晚期,发病超过7年,并有明显的躯干、四肢舞蹈症状,其中患者7已失去生活自理能力,此三者主要表现为明显的易激惹、发怒和攻击行为,均有刻板行为存在,1例(患者5)有明显的被害妄想(见表2)。本研究中HD患者最常见的症状为易激惹,晚期会有明显的妄想以及刻板思维、行为存在。
2.4神经影像学改变 本组HD的典型影像学检查结果为:对称性基底节萎缩,以双侧尾状核头部萎缩最为明显,导致侧脑室额角外侧面向外膨起,还可伴有尾状核的异常信号,并有全脑萎缩的改变(如图3A、D)。在本组已有症状的HD先证者中,影像学改变均符合HD典型的影像学改变。仅有一人(先证者11)不具备典型的头颅磁共振的改变,出现典型的舞蹈症状但是影像学检查改变仅可见明显的小脑萎缩,轻微尾状核萎缩(图3B、C)。
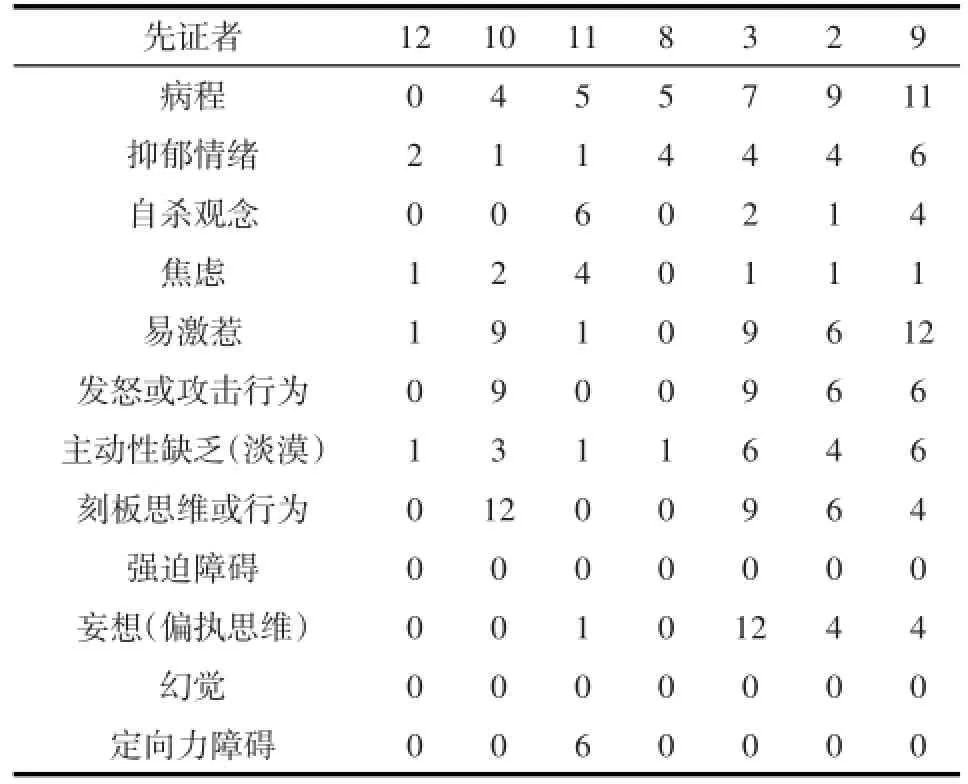
表2 HD患者行为学改变量表评估
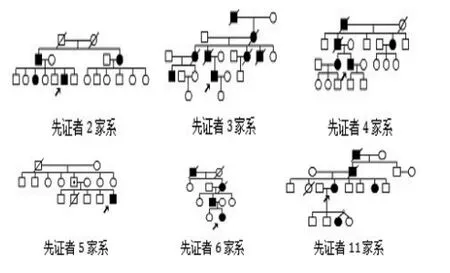
图2:部分有HD家族史家谱图展示
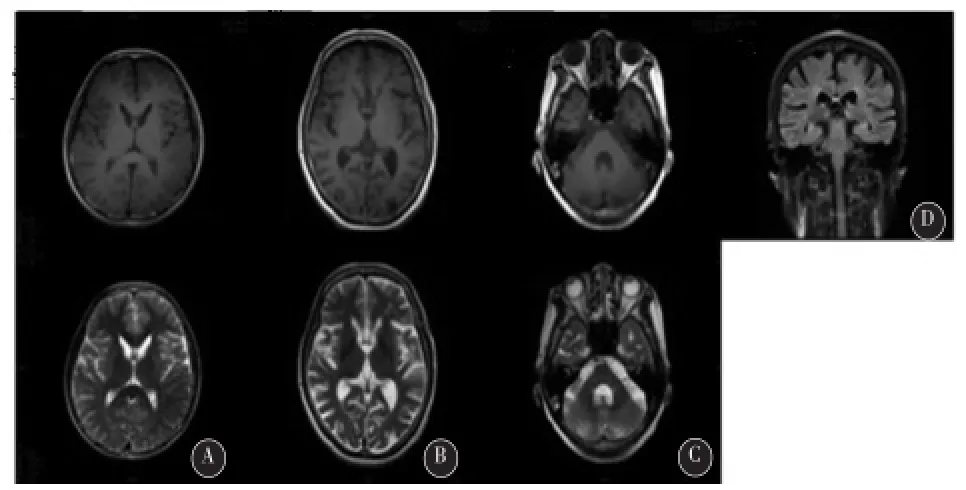
图3典型以及非典型HD颅脑MRI表现 A(先证者5):为典型的早期HD患者的MRI结果,可见明显的对称性双侧尾状核萎缩以及侧脑室的扩大;B,C(先证者11)为特殊HD患者的MRI结果,表现尾状核萎缩不明显,有明显的小脑萎缩;D(先证者9)典型的HD患者的MRI结果,可见全脑萎缩
3 讨论
HD为罕见的显性遗传的神经系统退行性疾病,系由于特异性的基底节及大脑皮质神经元的退行性病变导致的疾病。目前缺乏有效的治疗方法,由于HD症状的多样性,误诊常见,可被误诊为脊髓小脑共济失调(SCA)和肌张力障碍等[9],增加HD准确临床诊断的难度。因此,如何准确的早期诊断亨廷顿舞蹈症,寻找可以作为HD临床早期诊断或者病程发展评估的功能、神经化学等生物学标志物显得极为重要[10]。
对本组患者病史研究发现。除JHD患儿外,所有HD患者均以头面部、四肢、躯干的舞蹈样动作作为首诊的主诉。成年人HD临床最主要的症状为特征性的舞蹈样动作,多由头面部或四肢起始进而发展到全身,并呈进行性加重。同时在神经影像学方面,特殊的(MRI、CT)的表现为疾病诊断的重要参考依据之一,主要表现为特征性的基底节萎缩,以尾状核头部萎缩最明显,双侧侧脑室前角扩大[11-12]。阳性家族史是HD诊断的重要依据,但却非必要条件。由于追访不全或者其亲代与子代发病年龄差别等因素,往往会有假阴性家族史的存在。故而,临床结合其症状、影像学改变,如有阳性家族史需高度怀疑此遗传病,如有阴性家族史也需警惕HD的可能性。最终,基因诊断为该病的确诊检查手段。
本研究同时发现,虽然所有患者以运动症状作为首诊症状,其非运动障碍症状往往早于运动障碍出现[13-14]。本组患者中,6/10先证者在运动障碍出现之前已有行为学改变,并且以易激惹最常见。而7例接受了PBA问卷患者的研究中发现,所有患者(包括未病患者)均会出现不同程度的易激惹现象,运动症状出现前即存在并且随着运动症状加重而加重,并且可能进展为发怒以及攻击行为;抑郁、淡漠少见或较轻微。而国外对于HD患者行为学障碍临床随访调查研究数据显示[15-16]:HD患者主要有抑郁(>55%)、淡漠(~30%)以及易激惹(~10%)症状,且其严重程度的增加与运动症状的进展呈一定正相关。故而,就目前而言,行为学的研究似乎是可以作为疾病发生的一个提示性症状,并且在疾病进展过程中可作为评估指标之一。另外,本研究初步发现,中国HD患者的行为学改变与国外患者现有报道的症状不完全相同,或许提示中外HD发病症状的异质性。但对于行为学的改变是否可以作为疾病的生物学指标对疾病的发生、发展进行预测、评估,如何使用量表更加敏感的发现患者行为学的改变,仍需进一步的观察以及研究。
综上所述,HD临床诊断主要根据症状的三主征,但早期症状呈多样化,常以情绪、性格、行为学改变为先,确诊需要基因诊断协助。而在未来疾病的临床诊断中,还有望引入影像学方面已有相关结构(如尾状核头部最大直径、壳核最大直径、双额角比率;双尾状核比率[11-12])的量化指标作为参考。目前而言,HD缺少有效的生物学指标对疾病的发病进行预测或评估,故而,除典型的运动症状改变外,行为学改变以及影像学的变化有可能为早期HD临床诊断提供依据。由于现暂时未有有效的可以延缓或者终止疾病发生、发展的治疗方法,因此,早期对疾病进行监控,对患者的运动症状进行药物控制以及康复干预,对于患者生活质量的提高有重要意义。
[1]The Huntington's Disease Collaborative Research Group.A novel gene containing a trinucleotide repeats that is expanded and unstable on Huntington's disease chromosomes[J].Cell,1993,72(6):971-983.
[2]VONSATTEL JP,DIFIGLIA M.Huntington disease[J].Journal of neuropathology and experimental neurology,1998,57(5):369-384.
[3]WALKER FO.Huntington's disease[J].Lancet,2007,369(9557):218-228.
[4]GEEVASINGA N,RICHARDS FH,JONES KJ,et al.Juvenile Huntington disease[J].Journal of Paediatrics and Child Health,2006,42:552-554
[5]中华医学会神经病学分会帕金森病及运动障碍学组.亨廷顿病的诊断与治疗指南[J].中华神经科杂志,2011,44:638-641.
[6]Unified Huntington's Disease Rating Scale:reliability and consistency.Huntington Study Group[J].Movement disorders:official journal of the Movement Disorder Society,1996,11(2):136-142.
[7]ROSS CA,AYLWARD EH,WILD EJ,et al.Huntington disease:natural history,biomarkers and prospects for therapeutics [J].Nature reviews Neurology,2014,10(4):204-216
[8]KREMER B,SQUITIERI F,TELENIUS H,et al.Molecular analysis of late onset Huntington's disease[J].Journal of medical genetics,1993,30(12):991-995.
[9]POTTER NT,SPECTOR EB,PRIOR TW,et al.Technical standards and guidelines for Huntington disease testing[J].Genetics in medicine:official journal of the American College of Medical Genetics,2004,6(1):61-65.
[10]JEAN-MARCBURGUNDER,冯璐扬.亨廷顿舞蹈病:教学型综述[J].中国神经精神疾病杂志,2015,41(10):577-591.
[11]张宝荣,殷鑫浈,夏昆,等.舞蹈病家系临床、影像学特征及基因突变分析[J].中华神经科杂志,2005,38(11):686-689.
[12]田士强,张宇清,李勇杰.磁共振成像在亨廷顿病诊断中的应用价值[J].中国神经精神疾病杂志,2002,28(6):430-430,435.
[13]VANDUIJN E,REEDEKER N,GILTAY EJ,et al.Course of irritability,depression and apathy in Huntington's disease in relation to motor symptoms during a two-year follow-up period[J]. Neurodegenerative diseases,2014,13(1):9-16.
[14]左伋,夏蓓莉,陈秀珍,等.中国人Huntington舞蹈病发病情况的初步研究[J].中国神经精神疾病杂志,1994,20(4):221-222.
[15]PAULSEN JS,LONG JD,JOHNSON HJ,et al.Clinical and Biomarker Changes in Premanifest Huntington Disease Show Trial Feasibility:A Decade of the PREDICT-HD Study[J].Frontiers in aging neuroscience,2014,6:78.
[16]TABRIZI SJ,SCAHILL RI,OWEN G,et al.Predictors of phenotypic progression and disease onset in premanifest and early-stage Huntington's disease in the TRACK-HD study:analysis of 36-month observational data[J].The Lancet Neurology,2013,12(7):637-649.
(责任编辑:李立)
The clinical analysis of Huntington disease:a case study from 12 genetic diagnosis families.
SU Fengjuan,ZENG Yixuan,PEI Zhong,LIANG Xiuling,LI Xunhua,Jean-Marc Burgunder.Chinese Huntington’s Disease Network The First Affiliated Hospital of Sun-Yat sen University,Guangzhou 510080,China.Tel:020-87755766.
R742.2
A
10.3969/j.issn.1002-0152.2016.01.002
☆国家自然科学基金项目(编号:81171070)
*中山大学附属第一医院神经内科(广州 510080)
△中国亨廷顿病协作网(Chinese Huntington Disease Network)
◎瑞士伯尔尼大学神经科(University of Bern,Switzerland);欧洲亨廷顿病协作网(EuropeanHuntingtonDisease Network)
(E-mail:putaolaoda@sina.com)
2015-02-11)
猜你喜欢
广西林业科学(2022年6期)2023-01-16
中华实用诊断与治疗杂志(2022年1期)2022-08-31
中华实用诊断与治疗杂志(2022年1期)2022-08-31
临床输血与检验(2022年3期)2022-06-22
山东医药(2021年24期)2021-09-01
诊断学(理论与实践)(2020年1期)2020-04-28
科学24小时(2019年4期)2019-06-10
郑州大学学报(医学版)(2019年3期)2019-06-03
医药前沿(2019年29期)2019-01-05
科学生活(2016年7期)2016-07-25
