
北京鸭腹部脂肪组织的转录组特征分析
2016-06-16 01:07:47李国勤沈军达陶争荣卢立志
浙江农业学报 2016年5期
陈 黎,李国勤,田 勇,沈军达,陶争荣,徐 坚,曾 涛,卢立志
(浙江省农业科学院 畜牧兽医研究所,浙江 杭州310021)
北京鸭腹部脂肪组织的转录组特征分析
陈黎,李国勤,田勇,沈军达,陶争荣,徐坚,曾涛,卢立志*
(浙江省农业科学院 畜牧兽医研究所,浙江 杭州310021)
摘要:鸭的基因组序列虽已释放,但其基因组信息,尤其是转录组信息仍需进一步开发。文章利用转录组测序分析了鸭的腹部脂肪组织转录组特征。共获得203 200 984个高质量测序数据,鉴定出18 464个基因表达(RPKM≥1),其中96.9%的基因RPKM值小于1 000。15 070个基因发生了可变剪切,剪切次数为35 913次。统计可变剪切类型发现,内含子保留所占比例最低,占所有可变剪切类型的1.17%,而第一外显子可变剪切、末端外显子可变剪切、外显子跳跃依次是3种比例最高的可变剪切类型,比例分别为45.92%, 43.67%和6.23%。此外,利用这批转录组数据共检测出229 276个SNPs,其中转换是最主要的突变类型,占所有SNPs的73.28%。对SNP所在基因进行功能注释(GO)发现,这些基因涉及细胞组分、分子功能、生物学过程3大功能类别中广泛的生物功能,表明该研究开发的SNPs较为全面;通路分析(KEGG)发现,SNPs所在基因除了富集于脂类、能量代谢相关通路,更多的基因则富集于癌症、免疫以及内分泌系统相关的通路上,表明脂肪组织除了是能量储备组织,同时也是重要的免疫、内分泌组织。这些数据拓展了鸭的遗传信息,建立的SNPs数据库将有助于鸭分子标记辅助育种及功能基因定位。与癌症、免疫相关的SNPs可为癌症及免疫学研究提供候选遗传标记。
关键词:基因表达;可变剪切;单核苷酸多态性;脂肪组织
转录组测序(RNA-seq)是基于二代测序技术的转录组学研究方法。RNA-seq可在全基因组范围内检测转录本表达水平,同时还能发现未知转录本和新基因,识别可变剪切位点以及SNP(single nucleotide poly morphism)[1]。与传统的RNA研究手段(例如芯片)相比,RNA-seq具备灵敏度高、重复性好等优势。目前,随着测序技术发展及成本降低,RNA-seq已经成为转录组学研究的有效手段,借助该技术,多个物种的转录组信息得以释放。
鸭是重要的经济水禽,同时也是免疫研究的模式动物。目前,在鸭中已有利用RNA-seq开展转录组研究的报道。Li等[2]通过对鸭羽毛组织转录组测序,筛选出与羽色相关的候选基因;Tang等[3]对鸭肝脏组织的转录本进行了组装、注释,筛选出一批受甲肝病毒感染后表达变化的基因,为后续研究甲肝病毒感染的分子机制提供了线索。随后,Huang等[4]在公布鸭基因组序列的同时,利用RNA-seq检测了肺部被禽流感病毒感染后的基因表达变化。以上这些研究仅关注转录本的表达变化,尚未对可变剪切、SNP等信息进行报道。尽管Chen等[5]利用RNA-seq,比较了北京鸭与绿头野鸭在基因表达、可变剪切以及SNP方面的差异,但缺乏对鸭基因表达、可变剪切以及SNP的特征分析。 因此,本研究利用北京鸭腹部脂肪组织RNA-seq数据,对鸭脂肪组织内基因的表达水平及可变剪切特征进行分析,同时利用转录组数据构建SNP数据库,为鸭提供可用的遗传标记。
1材料与方法
1.1试验材料
1.1.1试验动物
本研究所用北京鸭来自湖州卓旺养鸭场。试验用鸭采用玉米豆粕型饲粮,自由采食和饮水。喂养至8周龄时进行屠宰,并立即取其腹部脂肪组织待提取RNA用。实验动物屠宰标准按照国家实验动物处理行为准则执行。
1.1.2主要试剂
提取RNA所用TRIzol试剂购自Life Technologies公司。
1.2试验方法
1.2.1RNA-seq测序、数据组装及注释
3只8周龄北京鸭,每只个体取300 mg腹脂,按照RNAeasy Lipid Tissue Mini Kit (QIAGEN,Germany)说明书分别提取总RNA。每个个体总RNA单独建库,随后利用Illumina HiSeq 2000 (Illumina, America) 平台进行双末端测序。测序数据过滤掉低质量数据及无用序列,包括测序接头、rRNA、tRNA以及miRNA等非编码RNA序列。获得高质量数据后,利用TopHat v2.0.9[6]将数据比对到鸭参考基因组上(BGI_duck_1.0),随后利用Cufflinks v2.1.1[7]默认参数对比对上的序列进行组装、注释及表达量的计算。
1.2.2SNP的挖掘及可变剪切鉴定
以鸭基因组(BGI_duck_1.0)做参考序列,利用SAMtools 0.1.19[8]寻找SNPs(single nucleotide polymorphisms),并利用BEDTOOLS 2.17.0对SNP进行筛选。可变剪切参照Florea 等[9]的方法利用ASprofile 进行分析。所有软件参数设置为默认参数。
1.2.3基因注释
GO(gene ontology)注释来源于GO数据库(http://www.geneontology.org/);通路分析采用KEGG(kyoto encyclopedia of genes and genomes)数据库。
2结果与分析
2.1北京鸭腹部脂肪组织基因表达分析
共获得203 200 984个高质量的北京鸭腹脂转录组数据,3个样本的Q20均大于95%,GC含量均接近50%,表明测序数据质量较好(表1)。RPKM(reads per kilo bases per million reads)是每百万reads 中来自某一基因每千碱基长度的reads 数目,是目前最常用的基因表达水平估算方法[10]。本研究以RPKM≥1作为基因表达标准,在北京鸭腹部脂肪组织中共鉴定出18 464个基因表达,其中11 047个基因在基因组上已有注释。对不同RPKM区间的基因数量进行统计发现,多数基因的RPKM值在1 000以下,其中RPKM值在1~10的基因最多,为8 600个;其次是RPKM值位于10~100的基因,数量为7 478个;RPKM值位于100~1 000的基因有1 806个,而RPKM值大于1 000的基因最少,有580个,占所有表达基因的3.1%。
表1测序数据统计表
Table 1Statistics of RNA-seq data
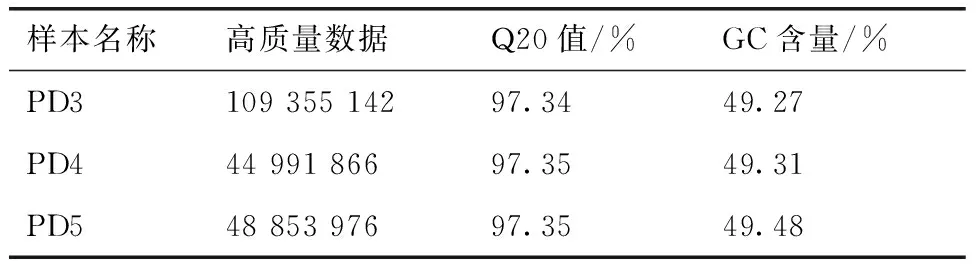
样本名称高质量数据Q20值/%GC含量/%PD310935514297.3449.27PD44499186697.3549.31PD54885397697.3549.48
注:PD3,PD4,PD5分别代表北京鸭的3个生物学重复。下同。
2.2北京鸭可变剪切分析
可变剪接是调节基因表达和产生蛋白质多样性的重要机制。本研究分析了5种主要的可变剪切类型,包括内含子保留(intron retention, IR)、5’或3’可变剪切(alternative exon ends, AE)、外显子跳跃(skipped exon, SE)、第一外显子可变剪切(alternative first exons, AF)、末端外显子可变剪切(alternative last exons, AL)。利用转录组数据,本研究共鉴定出约35 913次可变剪切,这些可变剪切由15 070个基因产生。在这5种可变剪切类型中IR所占比例最低,AF及AL比例最高,分别占所有可变剪切事件的45.92%及43.67%(表2)。
表2可变剪切类型统计表
Table 2Statistics of the alternative splicing (AS) events
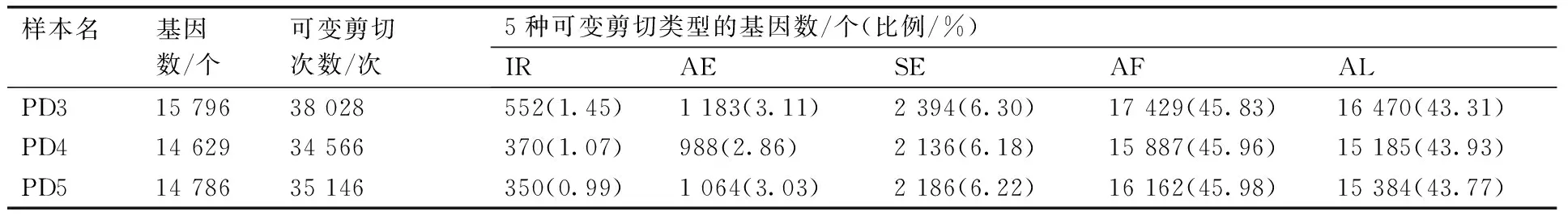
样本名基因数/个可变剪切次数/次5种可变剪切类型的基因数/个(比例/%)IRAESEAFALPD31579638028552(1.45)1183(3.11)2394(6.30)17429(45.83)16470(43.31)PD41462934566370(1.07)988(2.86)2136(6.18)15887(45.96)15185(43.93)PD51478635146350(0.99)1064(3.03)2186(6.22)16162(45.98)15384(43.77)
2.3SNP鉴定
利用SAMtools,共挖掘出229 276个SNP,其中168 028个为转换,61 248个为颠换。转换发生比例为73.28%,是最主要的突变类型(图1)。
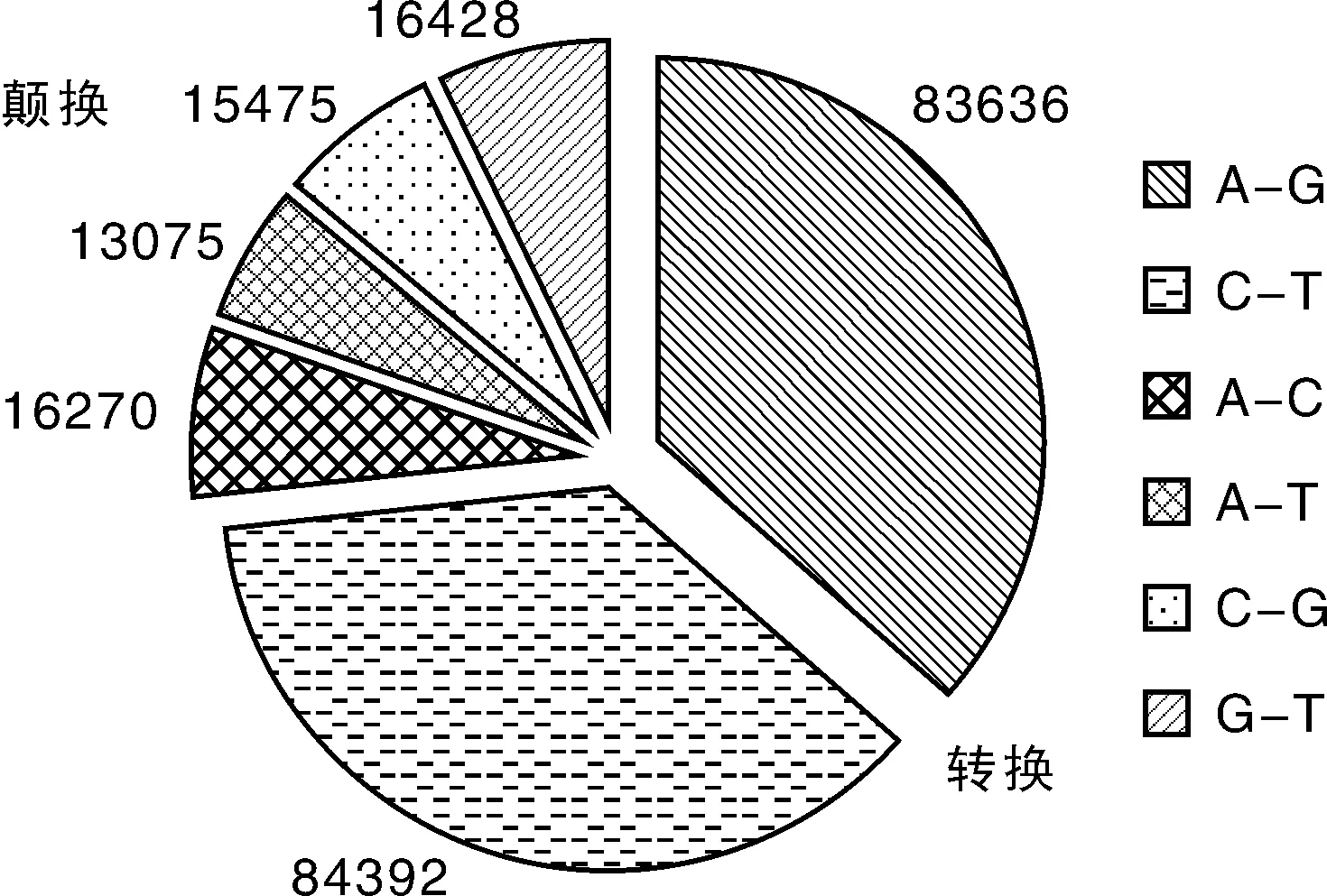
图1 SNPs类型统计Fig.1 Statistics of types of SNPs
GO注释发现SNP所在基因共归入到9 814个GO term中。根据富集的基因数量,分别统计分子功能、生物学过程、细胞组件中位于前10位的GO term(图2)。在分子功能中,与蛋白绑定(protein binding)、ATP绑定(ATP binding)以及锌离子绑定(zinc ion binding)相关的基因最多,分别为5 262,798,653个;在细胞组件中,位于细胞核(nucleus)、细胞质(cytoplasm)和细胞膜(membrane)的基因最多,分别有2 151,2 017和904个;生物学过程中,归入DNA依赖的转录调控(regulation of transcription, DNA-dependent)、RNA聚合酶Ⅱ启动子的转录正调控(positive regulation of transcription from RNA polymerase Ⅱ promoter)以及信号转导(signal transduction)3个GO term的基因最多,分别有473,401,392个。
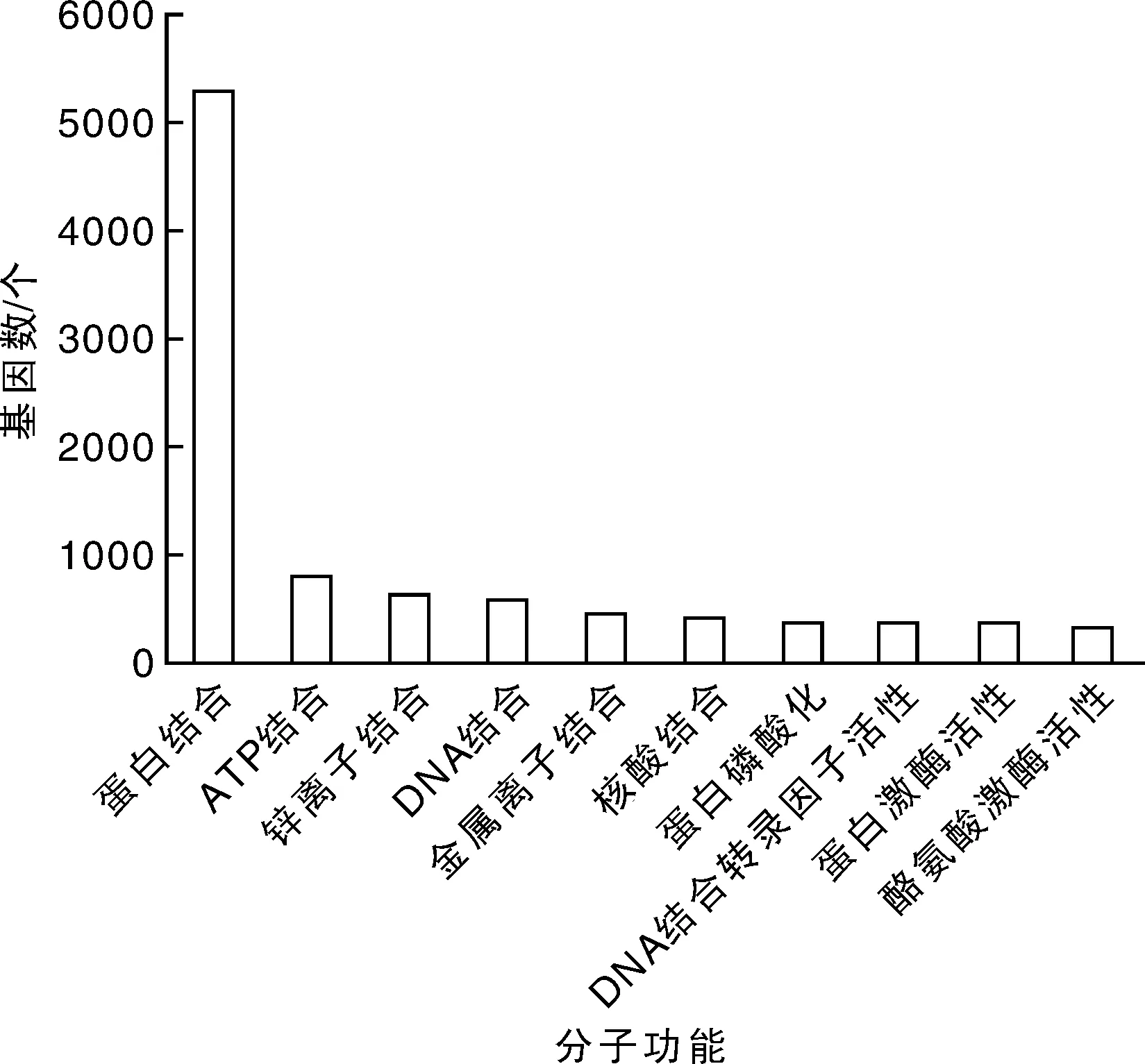
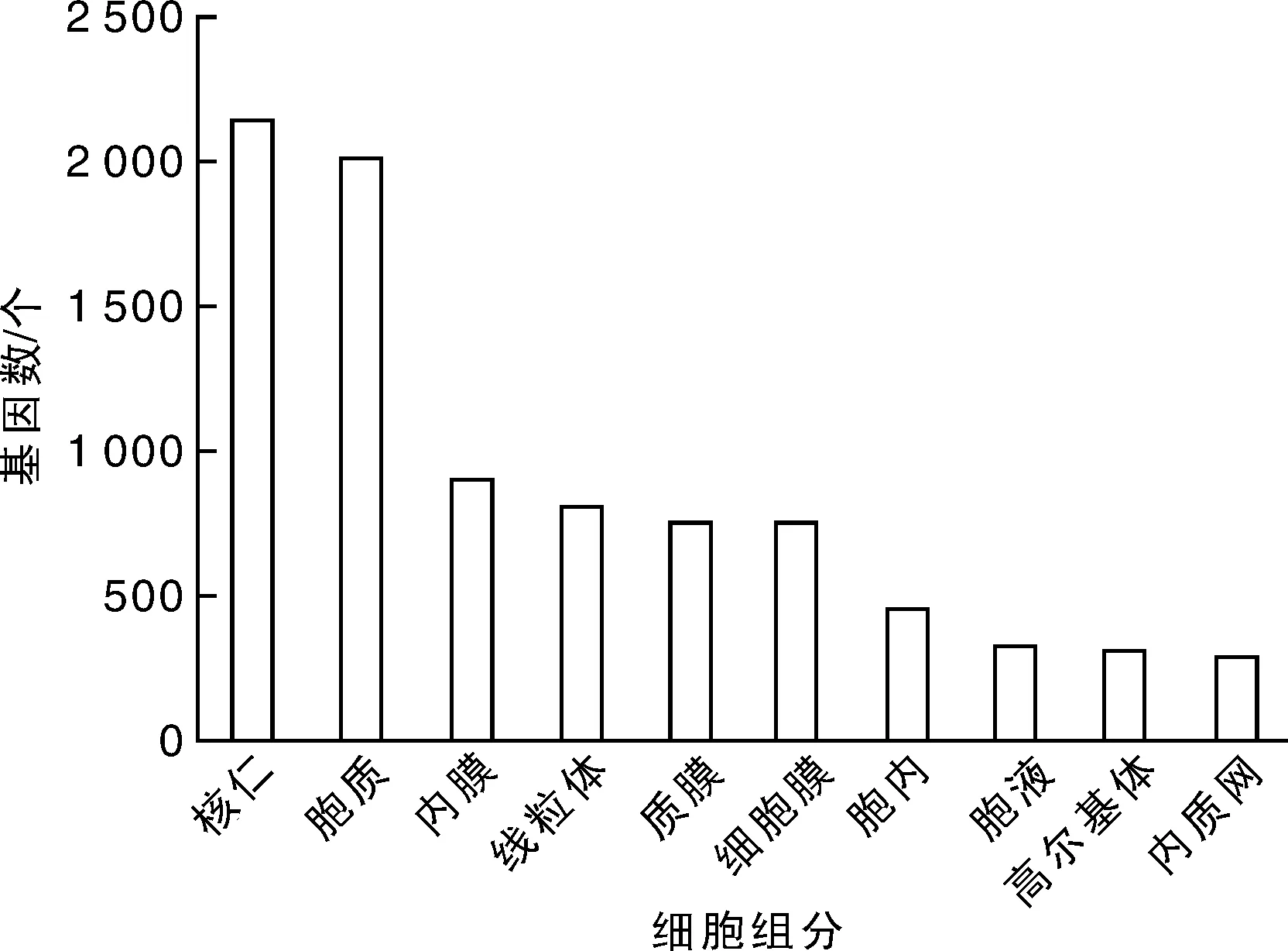
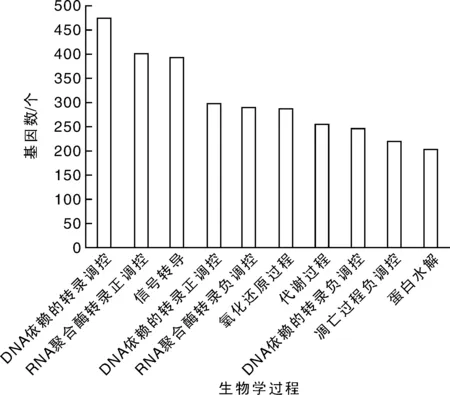
三大类别分别列出了富集基因数量位于前10位的GO term。图2 SNP所在基因的GO分类Fig.2 GO classifications of genes containing SNPs
同时,为确定SNP参与的主要生化代谢途径和信号通路,对SNP所在基因进行KEGG(kyoto encyclopedia of genes and genomes)通路分析,共鉴定出25个通路(图3),其中归入癌症的基因最多,为1 031个,其次是信号转导及免疫系统,分别有956个及812个基因;归入内分泌系统及脂肪代谢的基因也较多,分别有466和235个;归入碳水化合物代谢及能量代谢中的基因分别有138和66个。
3讨论
RNA-seq原始数据需要进行严格的质控。本研究在获得鸭转录组测序数据后,首先对原始数据进行质控。Q20与GC含量是衡量测序质量的重要指标。Q20表示质量数大于20的碱基占总体碱基的比例,Q20越高表明测序错误率低于1%的碱基比例越高。GC含量则用于分析建库或测序过程是否带来碱基偏离,正常情况下,4种碱基的出现频率接近。本研究3个样本的Q20及GC%的平均值分别为97.35%和49.35%,表明测序质量较高,为后续数据分析的可靠性奠定了基础。
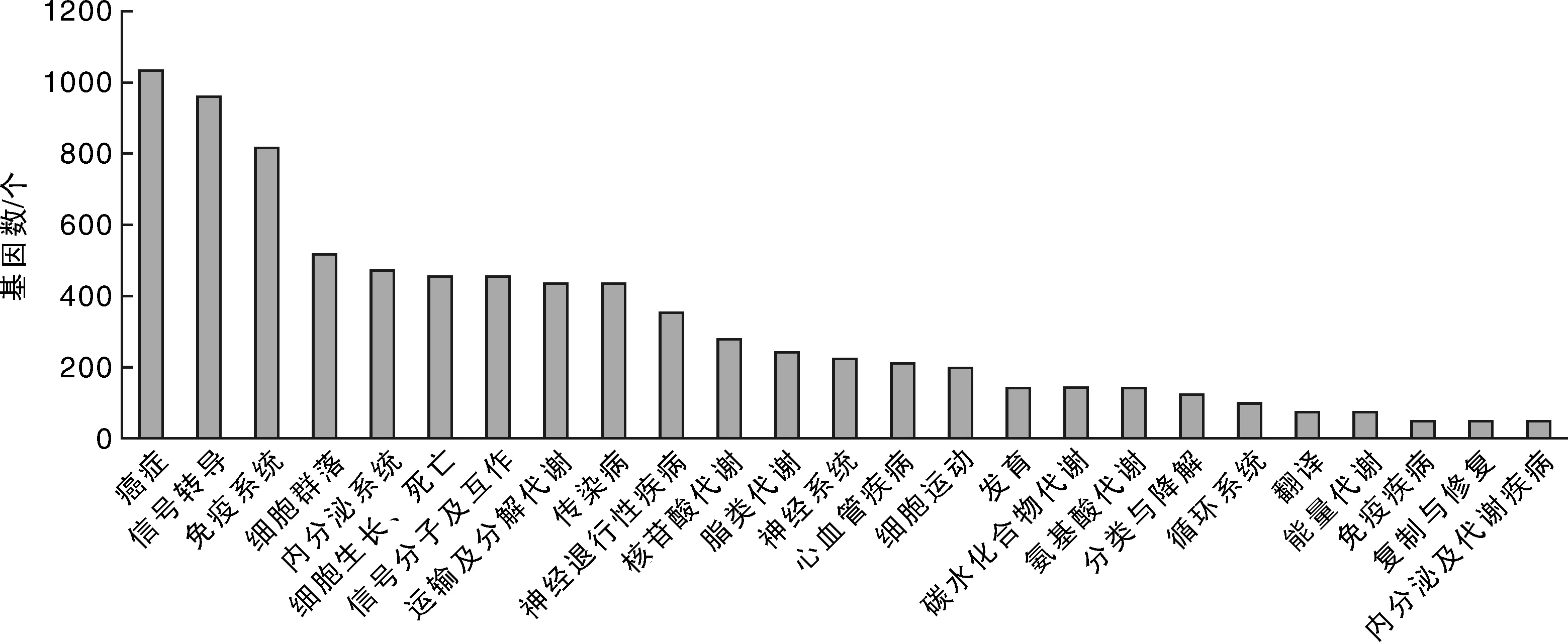
图3 SNPs所在基因的KEGG通路分析Fig.3 KEGG pathways of genes including SNPs
可变剪接是真核生物基因转录后调控的重要机制。基因通过可变剪切产生不同的转录本,使遗传信息得到放大。最近,Chen等[5]利用RNA-seq在鸭中鉴定出6 980个基因发生了23 393次可变剪切,发生可变剪切的基因数量远低于本研究中鉴定出的基因数量,这可能是因为可变剪切分析方法与鉴定标准不同。本研究利用北京鸭腹脂转录组数据共鉴定出18 464个基因表达,其中15 070个基因发生了35 913次可变剪切,可变剪切发生率约为82%,低于人类的95%[11],高于果蝇的60%[12],与越是高等生物可变剪切比例越高的报道一致[13]。在本研究中,IR在所有可变剪切类型中比例最低,与目前报道的动物中IR是比例最低的可变剪切类型[13-15]的观点一致。
利用转录组数据挖掘SNP已有多篇报道。Chen等[5]利用转录组测序比较了北京鸭与绿头野鸭腹部脂肪组织中的差异SNP,但未对全基因组SNP进行特征分析。本研究利用北京鸭腹部脂肪组织转录组数据共检测出229 276个SNP,这是目前鸭中首次在全基因组范围内挖掘SNP的报道,对SNP所在基因进行功能注释,发现这些基因涉及细胞组分、分子功能及生物学过程3大功能类别中广泛的生物功能,表明本研究开发出的SNP较为全面,与多种生物功能关联,方便了后续的基因定位及分子标记育种。对SNP所在基因通路分析发现,归入癌症以及免疫、内分泌系统的基因最多,这与脂肪组织除了是能量储备组织,同时也是重要的免疫、内分泌组织的报道相符[5,16],这些SNP可作为癌症、免疫相关的候选SNP。
参考文献:
[1]AN J, WAN H, ZHOU X, et al. A comparative transcriptomic analysis of uveal melanoma 465 and normal uveal melanocyte [J].PloSOne, 2011, 6(1): e16516.
[2]LI S, WANG C, YU W, et al. Identification of genes related to white and black plumage formation by RNA-Seq from white and black feather bulbs in ducks[J].PloSOne, 2012, 7(5): e36592.[3]TANG C, LAN D L, ZHANG H R, et al. Transcriptome analysis of duck liver and identification of differentially expressed transcripts in response to duck hepatitis A virus genotype C infection [J].PloSOne, 2013, 8(7): e71051.
[4]HUANG Y H, LI Y R, BURT D W, et al. The duck genome and transcriptome provide insight into an avian influenza virus reservoir species [J].NatureGenetics, 2013, 45(7): 776-783.[5]CHEN L, LUO J, LI J X, et al. Transcriptome analysis of adiposity in domestic ducks by transcriptomic comparison with their wild counterparts [J].AnimalGenetics, 2015, 46(3): 299-307.[6]TRAPNELL C, PACHTER L, SALZBERG S L. TopHat: discovering splice junctions with RNA-seq [J].Bioinformatics, 2009, 25(9): 1105-1111.
[7]TRAPNELLC, WILLIAMS B A, PERTEA G, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation [J].NatureBiotechnology, 2010, 28: 511-515.
[8]LI H, HANDSAKER B, WYSOKER A, et al. The sequence alignment/map (SAM) format and SAM tools [J].Bioinformatics, 2009, 25: 2078-2079.
[9]FLOREA L, SONG L, SALZBERG S L. Thousands of exon skipping events differentiate among splicing patterns in sixteen human tissues [J].F1000Research, 2013, 2: 188.
[10]MORTAZAVI A, WILLIAMS B A, MCCUE K, et al. Mapping and quantifying mammalian transcriptomes by RNA-Seq [J].NatureMethods, 2008, 5(7): 621-628.
[11]PAN Q, SHAI O, LEE J L, et al. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing [J].NatureGenetics, 2008, 40(12): 1413-1415.
[12]GRAVELEY B R, BROOKS A N, CARLSON J W, et al. The developmental transcriptome ofDrosophilamelanogaster[J].Nature, 2011, 471(7339): 473-479.
[13]KIM E, MAGEN A, AST G. Different levels of alternative splicing among eukaryotes [J].NucleicAcidsResearch, 2007, 35(1): 125-131.
[14]REDDY A S, ROGERS M F, RICHARDSON D N, et al. Deciphering the plant splicing code: experimental and computational approaches for predicting alternative splicing and splicing regulatory elements [J].FrontiersinPlantScience, 2012, 3(9): 18.[15]SUGNET C W, KENT W J, ARES M J, et al. Transcriptome and genome conservation of alternative splicing events in humans and mice [J].PacificSymposiumonBiocomputing, 2004: 66-77.
[16]MINER J L. The adipocyte as an endocrine cell [J].JournalofAnimalScience, 2004, 82(3): 935-941.
(责任编辑卢福庄)
Transcriptome analysis of abdominal fats from Peking ducks by RNA-seq
CHEN Li, LI Guo-qin, TIAN Yong, SHEN Jun-da, TAO Zheng-rong, XU Jian, ZENG Tao, LU Li-zhi*
(InstituteofAnimalHusbandryandVeterinaryScience,ZhejiangAcademyofAgriculturalSciences,Hangzhou310021,China)
Abstract:Although the duck genome sequence has been released, its genomic structure, especially its transcriptome characterization needs to be further studied. This study characterized the transcriptome of abdominal fat in ducks by using RNA-seq. In total, 203 200 984 clean reads were obtained, and 18 464 genes were identified to be expressed in abdominal fat, among which 96.9% genes’ RPKM values were lower than 1 000. 15 070 genes had alternative splicing (AS), and the splicing times were 35 913. Intron retention was found to be the rarest AS type, while alternative first exons, alternative last exons and skipped exon were the three major types, with the proportions of 45.92%, 43.67% and 6.23%, respectively. Using these transcriptome data, 229 276 SNPs were called, among which transitions were the main type, accounting for 73.28% of all the SNPs. Gene ontology(GO) annotation analysis revealed that genes including these SNPs took part in numerous functions belonging to the three categories of cellular component, molecular function, and biological process, and further kyoto encyclopedia of genes and genomes(KEGG) pathway analysis showed that most of these genes were related to cancer, immune system, and endocrine system, suggesting that the adipose tissue was an important immune and endocrine tissue, as well as an energy store tissue. These data could enlarge the genetic information of ducks. The numerous SNPs identified in this study would contribute to the molecular breeding and help in mapping the genes associated with important economic traits. The SNPs related with cancers and immune systems would serve as useful markers in related studies.
Key words:gene expression; alternative splicing; single nucleotide polymorphism; adipose tissue
基金项目:国家自然科学基金项目(31402065);浙江省自然科学基金项目(LQ14C170003)
作者简介:陈黎(1984—),女,江苏徐州人,博士,助理研究员,从事家禽遗传育种方面的研究。E-mail: chenli0429@163.com
*通信作者,卢立志,E-mail:lulizhibox@163.com
DOI:10.3969/j.issn.1004-1524.2016.05.05
中图分类号:S834+.81;Q789
文献标志码:A
文章编号:1004-1524(2016)05-0743-05
投稿日期:2016-01-28
浙江农业学报ActaAgriculturaeZhejiangensis, 2016,28(5): 743-747
http://www.zjnyxb.cn
陈黎,李国勤,田勇,等. 北京鸭腹部脂肪组织的转录组特征分析[J].浙江农业学报,2016,28(5): 743-747.
猜你喜欢
中老年保健(2021年9期)2021-08-24 03:49:52
昆明医科大学学报(2021年8期)2021-08-13 08:59:46
中国中药杂志(2017年1期)2017-03-06 21:19:22
中外医疗(2016年33期)2017-03-02 19:04:28
中国科技博览(2016年24期)2016-12-28 23:53:07
中国实用医药(2016年30期)2016-12-28 16:54:00
中国民族民间医药·上半月(2016年10期)2016-11-19 11:08:04
中国科技博览(2016年22期)2016-11-01 13:58:50
中国医药导报(2015年27期)2015-10-28 20:27:29
山东体育学院学报(2015年1期)2015-06-25 20:43:38
