
超高效液相色谱电喷雾串联四极杆质谱法检测豆制品中12种禁用工业染料
2016-05-13 09:32:04丁晓静杨蕴嘉
分析测试学报 2016年4期
赵 珊,张 晶,丁晓静,杨蕴嘉,邵 兵
(北京市疾病预防控制中心,食物中毒诊断溯源技术北京市重点实验室,北京 100013)
超高效液相色谱电喷雾串联四极杆质谱法检测豆制品中12种禁用工业染料
赵珊,张晶,丁晓静,杨蕴嘉,邵兵*
(北京市疾病预防控制中心,食物中毒诊断溯源技术北京市重点实验室,北京100013)
摘要:建立了超高效液相色谱-电喷雾串联四极杆质谱(UPLC-MS/MS)同时检测豆制品中12种橙黄色工业染料(碱性橙2、碱性橙21、碱性橙22、苏丹黄、苏丹橙G、二乙基黄、碱性嫩黄O、溶剂黄124、酸性橙Ⅱ、酸性间胺黄、酸性黄11和茜素黄R)的分析方法。样品经酸化乙腈提取;提取液于-20 ℃冷冻2 h后离心,可有效去除豆制品中的脂质类干扰物;梯度洗脱条件下经ACQUITY UPLC®BEH C18柱(1.7 μm,2.1 mm×100 mm)分离后采用多离子反应监测模式(MRM)检测,4种工业染料(酸性橙Ⅱ、酸性间胺黄、酸性黄11和茜素黄R)使用负离子模式,以乙腈-0.1%氨水为流动相,其余8种采用正离子模式检测,流动相为乙腈-0.1%甲酸水。结果表明:豆制品中12种工业染料的定量下限(LOQ)为0.2~10.0 μg/kg,3个加标水平的回收率为76.2%~122.0%,相对标准偏差为1.1%~7.4%,各项指标满足食品中痕量污染物检测的需要。该方法操作简便、灵敏度高,实现了12种禁用橙黄色染料的同时提取和净化,适合于豆制品中非法添加工业染料的筛查、确证。
关键词:超高效液相色谱-串联质谱;工业染料;豆制品
食品中非食用色素的违禁添加问题引起了广泛关注,2011年4月我国卫生部发布了《食品中可能违法添加的非食用物质和易滥用的食品添加剂名单》,在名单中涉及豆制品中可能违法添加的非食用色素有豆腐皮中添加碱性橙2(又名王金黄),豆制品中添加碱性嫩黄,豆瓣酱中添加酸性橙Ⅱ。为此,质监部门组织开展了豆制品及重点食品的专项检查,重点查处使用碱性橙2及其他非食用原料的违法行为。然而,豆制品中非法添加非食用色素的现象并未消失,2014年12月台湾媒体再度报道了豆制品中被查出添加工业染料二甲基黄(又名奶油黄、苏丹黄)、二乙基黄,这两种染料均为亲脂性偶氮化合物,被国际癌症研究署(IARC)列为2B等级的致癌物,禁止用作食品添加剂。为加强国内豆制品中违法添加的监管力度,扩充检测目标物,建立境内境外有可能添加的非食用色素同时筛查的方法是十分必要的。
食品中非法添加碱性橙2、碱性嫩黄O、酸性橙Ⅱ和酸性间胺黄等工业染料的检测方法已有报道,常见于液相色谱法[1-6]和液相色谱-串联质谱(LC-MS/MS)法[6-11];苏丹橙G、苏丹黄作为苏丹系列染料与苏丹红同时检测多采用LC-MS/MS法[ 7-8,12-14],近期豆制品中添加二甲基黄(苏丹黄)、二乙基黄的液相色谱-质谱联用法也已见文献报道[15-16],然而对于豆制品中碱性橙2、碱性橙21、碱性橙22、碱性嫩黄O、酸性橙Ⅱ、酸性间胺黄、苏丹橙G、苏丹黄和二乙基黄9种橙黄色目标物的同时检测方法,尚未见文献报道。本研究亦将脂溶性染料溶剂黄124和水溶性染料酸性黄11、茜素黄R作为豆制品中橙黄色染料检测的目标物,建立了豆制品中极性差异很大的12种橙黄色染料的同时提取、净化和LC-MS/MS检测方法。
1实验部分
1.1仪器、试剂与材料
ACQUITYTM超高效液相色谱仪、 Xevo TQ-S 三重四极杆串联质谱仪(Waters公司);Milli-Q超纯水器(美国Millipore公司);11BS25型粉碎机(德国IKA公司)。碱性橙2(C12H12N4·HCl,纯度98.2%)、碱性橙21(C22H23ClN2,99.5%)、碱性橙22(C28H27ClN2,100.0%)购于振翔公司;苏丹黄(二甲基黄)(C14H15N3,99.5%)、苏丹橙G(C12H10N2O2,98.6%)、二乙基黄(C16H19N3,99.0%)、酸性橙Ⅱ(C16H11N2NaO4S,94.0%)、溶剂黄124(C22H31N3O2,99.0%)、酸性黄11(C16H13N4NaO4S,100.0%)、酸性间胺黄(C18H14N3NaO3S,99.0%)购于Dr.Ehrenstorfer GmbH;碱性嫩黄O(C17H21N3·HCl,98.0%)、茜素黄R(C13H9N3O5,96%)购于Acros Organics公司。标准储备液均用乙腈溶解配制,按纯度标示折算制成100 mg/L。乙腈、甲醇(色谱纯,Dikma 科技有限公司);甲酸(98%,Acros Organics公司);氯化钠(分析纯,国药集团化学试剂有限公司)。
1.2样品预处理
称取1 g(精确至0.01 g)粉碎的豆制品样品于50 mL塑料离心管内,加入2.5 mL水、50 μL冰乙酸、10 mL乙腈,涡旋混匀15 s,超声萃取30 min,静置分层,4 ℃下以10 000 r/min离心10 min后,转移上清液于另一50 mL刻度管内,残渣再加入50 μL冰乙酸、10 mL乙腈提取1次,合并上清液。提取液中加入约0.5 g氯化钠,于-20 ℃冷冻2 h,取出后于4 ℃下 10 000 r/min离心5 min,乙腈层用于LC-MS/MS测定。
称取1 g空白基质样品,每份样品按上述萃取过程进行处理,得到的基质提取液用于配制基质匹配工作曲线及高浓度样品溶液稀释。
1.3色谱-质谱条件
1.3.1正离子测定色谱柱:ACQUITY UPLC®BEH C18(1.7 μm,2.1 mm×100 mm);柱温:40 ℃;流动相:A为乙腈;B为0.1%甲酸水溶液。流速:0.3 mL/min;进样量:5 μL。梯度洗脱程序:0~10.0 min,15%A线性升至100%A;保持2 min,然后迅速降至15%A。
ESI(+)离子源:毛细管电压:2.8 kV;离子源温度:100 ℃;脱溶剂气温度:350 ℃;脱溶剂气流量:800 L/h。
1.3.2负离子测定色谱柱同上;柱温:40 ℃;流动相:A为乙腈;B为0.1%氨水。流速:0.3 mL/min;进样量:5 μL。梯度洗脱程序:0~1.5 min,10% A;1.5~5.0 min线性升至100%A;保持2 min,然后迅速降至10%A。
ESI(-)离子源:毛细管电压:2.5 kV;离子源温度:100 ℃;脱溶剂气温度:350 ℃;脱溶剂气流量:800 L/h 。各种染料的质谱采集参数见表1。
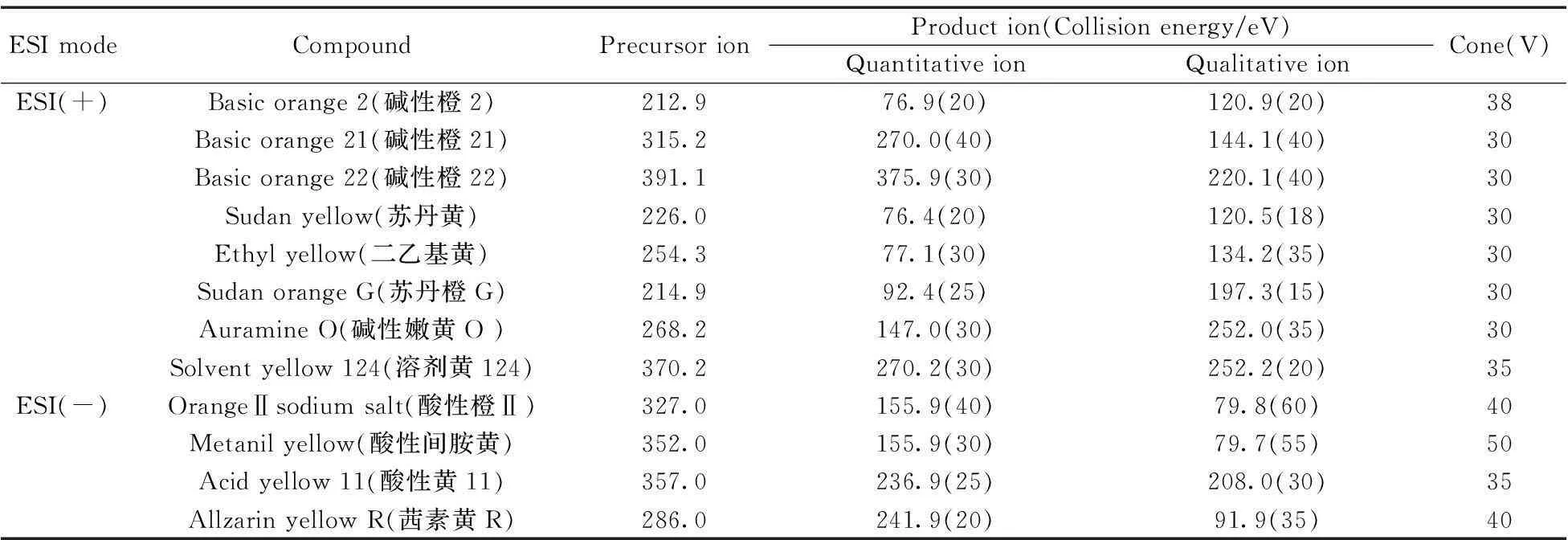
表1 12种染料的质谱采集条件
2结果与讨论
2.1液相色谱-质谱条件的优化
溶剂黄124、酸性黄11和茜素黄R 3种染料的检测未见文献报道,分别对其单标标准溶液进行一级离子扫描,确定溶剂黄124的电离方式为正离子模式,酸性黄11和茜素黄R为负离子模式。其中碱性橙2、碱性橙21、碱性橙22、苏丹黄、苏丹橙G、二乙基黄、碱性嫩黄O、溶剂黄124在ESI(+)检测模式下,酸性橙Ⅱ、酸性间胺黄、酸性黄11和茜素黄R在ESI(-)检测模式下均可获得两个丰度比较高的子离子,分别作为定性和定量离子,建立的多离子反应监测(MRM)模式的质谱采集参数见表1。
ESI(+)检测模式下的8种化合物极性差异较大,以0.1%甲酸水和乙腈为流动相,优化梯度洗脱条件,8种染料组分在ACQUITY UPLC®BEH C18色谱柱上获得了良好分离。豆制品中8种染料的加标色谱图见图1。ESI(-)检测模式下的4种化合物极性均较强,首先采用水-乙腈为流动相进行优化,茜素黄R的色谱峰形拖尾,进而采用10 mmol/L乙酸铵-乙腈、10 mmol/L碳酸铵-乙腈、0.1%氨水-乙腈优化分离条件,当流动相中加入少量乙酸铵或碳酸铵时均可使茜素黄R的色谱峰形得到改善,抑制拖尾;然而流动相中加入少量乙酸铵会使得酸性橙Ⅱ的响应降低,碳酸铵的加入使得酸性黄11的峰形展宽;而采用0.1%氨水-乙腈为流动相时,各组分的色谱峰形良好,负离子化效率明显增强,检测灵敏度提高。图2为豆制品基质中负离子模式下4种染料的加标色谱图。
2.2样品前处理方法的选择
2.2.1提取溶剂的选择12种目标化合物中酸性橙Ⅱ等4种负离子模式化合物和碱性橙2的极性强、水溶性好,而溶剂黄124、苏丹黄和二乙基黄的极性较弱、脂溶性好,因此提取溶剂的选择需考虑对各组分的溶解能力,乙腈为首选溶剂[8,14];对于脱水的豆制品样品如腐竹、豆皮等,若直接用乙腈提取,不易渗透到样品组织,因此需加入水,使其溶胀。本研究选择水-乙腈、水-乙酸-乙腈及水-硫酸铵-乙腈3种溶剂进行了提取率的考察,以基质匹配外标法计算,结果见图3。其中,水-硫酸铵-乙腈的提取效率相对较低,水-乙腈和水-乙酸-乙腈均具有较高的提取率,考虑到豆制品中含有30%~40%的蛋白质,酸性溶液有利于蛋白沉淀,因此选择水-乙酸-乙腈作为提取溶剂。考察了提取次数对回收率的影响,实验表明第2次提取各目标化合物的提取率在5.2%~17.8%之间。由此可见,两次提取才可获得较高的回收率。





2.2.2净化豆制品中含有的脂质类成分被萃取到提取液中,可能会对质谱响应产生抑制,降低灵敏度,并影响色谱柱的使用寿命。固相萃取是食品基质中目标化合物常用的净化手段,金玉娥等[3]采用WAX固相萃取小柱净化豆干和黄鱼中的酸性橙Ⅱ和酸性间胺黄,然而由于本研究中12种化合物的结构差异非常大,考虑到成本因素,很难采用固相萃取净化,因此采用盐析和冷冻的方式去除脂肪等干扰物质:即在提取液(乙腈及少量水)中加入氯化钠置于-20 ℃冷冻2 h,取出后离心,提取溶液分层,上层乙腈澄清透明,由此可见,冷冻后蛋白、脂肪沉淀析出,达到了净化提取液的目的。由图1~2可见,12种目标化合物经色谱分离,MRM模式进行检测后,色谱峰无杂质干扰。
2.3方法学验证
2.3.1标准曲线与定量下限取豆制品空白样品,加入混合标准系列,按“1.2”样品预处理方法进行提取、净化,以选定的定量离子峰面积(Y)对含量(X,μg/kg)绘制标准曲线,12种染料的线性范围及相关系数见表2。正离子模式下,碱性橙2、碱性橙21、碱性橙22、苏丹黄、苏丹橙G、二乙基黄、碱性嫩黄O、溶剂黄124 的线性范围为1.0~25.0 μg/L;负离子模式下,另外4种待测物的线性范围为2.5~62.5 μg/L。从样品加标提取液获得每种化合物的信噪比接近于10(S/N=10)的浓度,确定为该化合物的定量下限(LOQ)。由表2可见,12种染料的LOQ为0.2~10.0 μg/kg,表明样品经提取净化后,减少了基质抑制,可获得较高的灵敏度。

表2 豆制品中12种染料的线性范围、线性方程、相关系数及定量下限
2.3.2回收率与相对标准偏差分别取豆制品空白基质,添加3个浓度水平的混合标准溶液,按“1.2”方法进行处理,UPLC-MS/MS测定提取液中目标化合物的浓度。每个浓度点取6份样品进行平行实验,计算平均回收率和相对标准偏差(RSD),结果见表3。 12种染料的回收率为76.2%~122.0%,RSD为1.1%~7.4% 。说明该方法可获得较理想的提取效果。

表3 豆制品中12种染料的回收率和相对标准偏差(n=6)
2.4方法应用
应用本方法对市售25份样品(腐竹、豆皮、豆干)进行测定,未检出阳性样品。
3结论
本文建立了超高效液相色谱-电喷雾串联四极杆质谱(UPLC-MS/MS)检测豆制品中12种橙黄色工业染料的方法,样品经酸化乙腈提取、-20 ℃冷冻净化后可有效除去蛋白和脂质类干扰物,使得12种目标化合物达到同时提取净化。通过优化质谱条件,8种工业染料(碱性橙2、碱性橙21、碱性橙22、苏丹黄、苏丹橙G、二乙基黄、碱性嫩黄O、溶剂黄124 )采用正离子模式,乙腈-0.1%甲酸水为流动相;另4种工业染料(酸性橙Ⅱ、酸性间胺黄、酸性黄11和茜素黄R)采用负离子模式,以乙腈-0.1%氨水为流动相,均可获得较高的灵敏度和较好的峰形。UPLC-MS/MS检测方法的定量下限、回收率和精密度均满足痕量分析的要求,适用于豆制品中12种工业染料的同时筛查与确证。
参考文献:
[1]Lin Q.Chin.J.Chromatogr.(林钦.色谱),2007,25(5):776-777.
[2]Liu M,Li X L,Bie W,Wang M L,Feng Q.Chin.J.Chromatogr.(刘敏,李小林,别玮,王明林,冯骞.色谱),2011,29(2):162-167.
[3]Jin Y E,Sun P,Shen H,Wen Y M, Zhang H M,Wang G Q.ShanghaiMeas.Test.(金玉娥,孙丕,沈红,温忆敏,张慧敏,汪国权.上海计量测试),2006,(4):19-21.
[4]Zheng Y M,Guo W,Nie X M,Yang L,Pan J R,Chu X G.Chin.Agric.Sci.Bull.(郑月明,国伟,聂雪梅,杨玲,潘家荣,储晓刚.中国农学通报),2012,28(9):222-228.
[5]Zhao H Y,Zhao R,Li B,Chen Z H,Wu G H.Chin.J.FoodHyg.(赵海燕,赵榕,李兵,陈忠辉,吴国华.中国食品卫生杂志),2011,23(6):527-531.
[6]Chen L,Wen J X,Lei Y,Zhang R.J.Instrum.Anal.(陈林,温家欣,雷毅,张荣.分析测试学报),2015,34(9):1008-1013.
[7]Botek P,Poustka J,Hajšlová J.CzechJ.FoodSci.,2007,25:17-24.
[8]Lu Y,Qu Y,Feng N,Zhao J P, Jin W W.FoodSci.(路勇,渠岩,冯楠,赵俊平,金伟伟.食品科学),2012,33(6):176-180.
[9]Zhu X J,Yan C R,Dong C,Xu C X.J.FoodSafetyQuality(朱晓军,颜春荣,董璨,徐春祥.食品安全质量检测学报),2012,3(3):190-194.
[10]Cao P,Qiao X G,Lou X S,Geng J P,Fu J,Zhang X Q.Chin.J.Anal.Chem.(曹鹏,乔旭光,娄喜山,耿金培,付建,张禧庆.分析化学),2011,39(11):1670-1675.
[11]Lin D Q,Wan C B,Qiu P,Liu H M.J.Chin.MassSpectrom.(林黛琴,万承波,邱萍,刘花梅.质谱学报),2013,34(3):170-178.
[12]Sun H W,Wang F C,Ai L F.J.Chromatogr.A,2007,1164:120-128.
[13]Zhao S,Yin J,Zhang J,Ding X J,Wu Y N,Shao B.FoodAnal.Methods,2012,5:1018-1026.
[14]Ma Y S,Li W,Ai L F,Guo C H,Chen R C.J.Instrum.Anal.(马育松,李玮,艾连峰,郭春海,陈瑞春.分析测试学报),2014,33(1):93-97.
[15]Fan S F,Li Q,Ma J M,Li H,Zhang Y.Chin.J.Chromatogr.(范素芳,李强,马俊美,李挥,张岩.色谱),2015,33(6):657-661.
[16]Yan H,Yu T T,Jiang L P, Zhang L,Wang H X,Zhu X L,Li X.J.FoodSafetyQuality(严恒,余婷婷,蒋丽萍,张莉,王会霞,朱晓玲,黎星.食品安全质量检测学报),2015,6(4):1459-1462.
Determination of 12 Banned Dyes in Soy Products by Ultra Performance Liquid Chromatography with Electospray Ionization Tandem Mass Spectrometry
ZHAO Shan,ZHANG Jing,DING Xiao-jing,YANG Yun-jia,SHAO Bing*
(Beijing Key Laboratory of Diagnostic and Traceability Technologies for Food Poisoning,Beijing Center for Disease Prevention and Control,Beijing100013,China)
Abstract:An analytical method was developed for the simultaneous determination of 12 banned dyes(basic orange 2,basic orange 21,basic orange 22,dimethyl yellow,ethyl yellow,sudan orange G,auramine O,solvent yellow 124,orangeⅡsodium salt,metanil yellow,acid yellow 11 and allzarin yellow R) in soy products by ultra performance liquid chromatography-tandem mass spectrometry(UPLC-MS/MS).Soy products samples were extracted using acetonitrile acidified with acetic acid.After frozen for 2 h under-20 ℃,the extracts were centrifuged to reduce lipids and other related interference.The target compounds were separated on an ACQUITY UPLC®BEH C18column by means of a binary mobile phase gradient with water containing 0.1% ammonium hydroxide formic acid and acetonitrile for 4 dyes(orangeⅡsodium salt,metanil yellow,acid yellow 11 and allzarin yellow R) and 0.1% formic acid and acetonitrile for the other eight compounds.Mass spectrometric acquisition was carried out by means of multiple reaction monitoing(MRM) under negative ionization mode for 4 dyes and positive mode for the rest of target compounds,respectively.As a result,quantitation limits of 12 dyes in soy products samples were in the range of 0.2-10.0 μg/kg.Mean recoveries at three spiked levels ranged from 76.2% to 122.0%,and the relative standard deviations(RSDs)were between 1.1% and 7.4%,which complied with the regulations for the determination of trace contaminants residues in food matrix.Simultaneous extraction and purification of 12 dyes in soy products matrix was achieved upon the established method which could be applied in the routine detection of illegally additive dyes in soy products due to its simplicity and high sensitivity.
Key words:ultra performance liquid chromatography-tandem mass spectrometry(UPLC-MS/MS);dyes;soy products
收稿日期:2015-09-06;修回日期:2015-09-29
基金项目:质检总局公益性行业科研项目(2012104003-3)
*通讯作者:邵兵,博士,研究员,研究方向:食品安全分析,Tel:010-64407191,E-mail:shaobingch@sina.com
doi:10.3969/j.issn.1004-4957.2016.04.010
中图分类号:O657.63;F767.4
文献标识码:A
文章编号:1004-4957(2016)04-0432-06
猜你喜欢
中国生殖健康(2019年5期)2019-01-06 09:17:12
现代营销(创富信息版)(2018年7期)2018-09-05 03:24:32
现代营销(创富信息版)(2018年6期)2018-09-05 03:14:26
现代营销(创富信息版)(2018年5期)2018-07-12 01:11:02
中国中药杂志(2016年21期)2017-02-16 14:07:58
中国中药杂志(2016年21期)2017-02-16 13:02:24
分析化学(2017年1期)2017-02-06 21:34:35
分析化学(2017年1期)2017-02-06 21:32:17
热带农业科学(2016年10期)2016-12-12 01:52:56
上海医药(2016年1期)2016-02-22 10:32:47
