
潮霉素B抗性为选择标记的整合型表达载体的构建及在米根霉中的遗传转化
2015-12-27 07:27:44张旻姜绍通郑娟郑志李兴江潘丽军罗水忠
生物工程学报 2015年8期
张旻,姜绍通,郑娟,郑志,李兴江,潘丽军,罗水忠
潮霉素B抗性为选择标记的整合型表达载体的构建及在米根霉中的遗传转化
张旻,姜绍通,郑娟,郑志,李兴江,潘丽军,罗水忠
合肥工业大学生物与食品工程学院,安徽 合肥 230009

张旻, 姜绍通, 郑娟, 等.潮霉素B抗性为选择标记的整合型表达载体的构建及在米根霉中的遗传转化. 生物工程学报, 2015, 31(8): 1203–1218.Zhang M, Jiang ST, Zheng J, et al. Construction of an integration vector carrying hygromycin B resistance gene and its genetic transformation in Rhizopus oryzae. Chin J Biotech, 2015, 31(8): 1203–1218.
为了建立适合米根霉的遗传转化体系,应用重叠延伸PCR的方法构建了以潮霉素B抗性为选择标记的单交换整合型表达载体pBS-hygro-A;分别采用PEG/CaCl2介导的原生质体转化、原生质体电转化及萌发孢子电转化的方法将表达载体pBS-hygro-A转化入米根霉AS 3.819菌株中,并研究了菌丝酶解时间、孢子萌发时间以及电转化电场强度对于转化效率的影响;通过荧光定量PCR (qPCR) 对米根霉转化子基因组中质粒整合拷贝数进行了检测,并研究了其对米根霉转化子抗性稳定性的影响。实验结果表明成功获得整合了表达载体pBS-hygro-A的米根霉转化子。菌丝酶解140 min产生的原生质体其再生率和转化率最高,原生质体电转化最佳电场强度为13 kV/cm,孢子萌发2.5 h转化率最高,萌发孢子电转化最佳电场强度为14 kV/cm。萌发孢子电转化方法转化率要高于原生质体转化的方法。荧光定量PCR检测结果表明,在一定范围内,高质粒整合拷贝数的米根霉转化子比较稳定。研究建立了用于工业米根霉菌株的遗传转化体系,为米根霉代谢调控研究以及菌种改造工作提供了基础与支持。
米根霉,整合载体,原生质体,萌发孢子,同源重组,重叠延伸PCR,电穿孔
米根霉是一种重要的工业微生物,作为生产高纯度L-乳酸的理想菌种之一得到广泛应用[1-3]。由于其培养条件简单,具有高效的分泌表达系统以及发酵工艺成熟易于实现大规模连续发酵等优点,也有着应用于同源或者异源蛋白表达,成为一种高效基因工程菌的潜力。此外米根霉全基因组测序工作已经完成并已在互联网中进行了公布,许多有待研究的未知功能的基因,其序列及在基因组中所处位置也已为世人所知晓[4]。这也为米根霉基因改造工作提供了重要的前提条件。
在丝状真菌中,粗糙脉孢霉和构巢曲霉作为真菌遗传转化基础研究的重要材料得到了深入的研究及应用,并发展出了相对成熟的遗传转化操作方法与体系。此外,近年来围绕其他曲霉属及木霉属的遗传转化方面的研究也得到了长足发展。越来越多的丝状真菌已经作为基因操作宿主用于真核遗传转化体系的建立及高效基因工程菌的选育。然而对于米根霉遗传转化系统的研究却十分有限,且只适用于非常特殊的特定营养缺陷型米根霉菌株,而非广泛适用[5-10]。由于米根霉在工业生产中的重要性及其作为外源基因表达宿主所具有的优越性,对米根霉进行遗传改良是必然的趋势。
本文在前人研究基础之上,同样尝试了以整合入基因组为基础的载体构建策略,以近年来在丝状真菌遗传转化研究当中得到应用的潮霉素B抗性作为筛选标记,并加以适量的添加剂Triton X-100优化筛选结果。应用了重叠延伸PCR技术[11-13](也称融合PCR,gene splicing by overlap extension PCR,简称SOE PCR) 构建了适用于米根霉遗传转化的表达载体pBS-hygro,并在此载体上克隆了乳酸脱氢酶A基因A重组表达盒PAATA,构成重组载体pBS-hygro-A。应用PEG/CaCl2介导的原生质体化学转化、原生质体电转化及萌发孢子电转化转化的方法,将重组载体转化入米根霉中,筛选鉴定得到转化子,期望依此研究建立米根霉的高效遗传转化方法,为今后米根霉基因改良工作奠定基础。
1 材料与方法
1.1 材料
1.1.1 菌株
米根霉AS 3.819菌株购自中国工业微生物菌种保藏管理中心 (CICC No. 40313);大肠杆菌JM 109购自大连宝生物有限公司。
1.1.2 培养基及溶液
菌丝体液体培养基 (g/L):KH2PO40.15,MgSO4·7H2O 0.25,NaH2PO40.134,ZnSO4·7H2O 0.01,葡萄糖120,(NH4)2SO46。
菌丝酶解溶液:蜗牛酶、纤维素酶及溶壁酶以1∶1∶1的比例混合溶解于磷酸盐缓冲液 (pH 5.8,50 mmol/L) 配制的0.6 mol/L山梨醇溶液中,使酶总量终浓度为1.5 g/L。酶解溶液采用0.22 μm无菌滤膜过滤除菌。
YED培养基 (g/L):酵母提取物10,葡萄糖20,以20 mmol/L HEPES (pH 7.5) 溶液配制。
YED原生质培养基:YED培养基中添加山梨醇至终浓度为0.6 mol/L。
PDA液体培养基:称取200 g马铃薯,切碎加水1 000 mL煮沸0.5 h,纱布过滤,再加20 g葡萄糖充分溶解后趁热纱布过滤,并定容至1 000 mL。
PDA固体培养基:PDA液体培养基中增加琼脂至浓度为20 g/L,Triton X-100至0.05% (/)。
原生质体再生培养基:PDA液体培养基培养基中增加山梨醇至浓度为0.6 mol/L。
原生质体再生固体培养基:PDA固体培养基增加山梨醇至浓度为0.6 mol/L。
筛选培养基:PDA固体培养基中加入潮霉素B至120 μg/mL。
原生质体筛选培养基:原生质体再生固体培养基中加入潮霉素B至120 μg/mL。
1.1.3 质粒
pBC-hygro及pBluescript SK+质粒购自Biovector质粒载体菌株细胞株基因保藏中心 (Science Lab,Inc);pEASY-T1克隆载体购自北京全式金生物技术有限公司 (TransGen Biotech);本研究所用到的质粒均列于表1中。
1.1.4 酶和试剂
rDNA聚合酶、DNA聚合酶、DNA限制性内切酶Ⅰ、Ⅰ、Ⅰ、T4 DNA连接酶、DNA marker、IPTG、X-gal以及SYBR Premix ExⅡ荧光定量PCR酶均为TaKaRa公司产品。潮霉素 (Hygromycin B) 为Roche公司产品;氨苄青霉素 (Ampicillin) 为Amresco 产品。以上试剂均为分析纯和生化试剂。PCR产物胶回收纯化试剂盒为Axygen公司产品。质粒小量抽提试剂盒以及pEASY-T1克隆载体购自北京全式金生物工程有限公司。

表1 本研究所用质粒
1.1.5 仪器
TC-96 PCR仪 (杭州博日科技有限公司),Universal HoodⅡ凝胶成像系统 (Bio-Rad),光学显微镜 (Zeiss),Multiporator电转化仪 (Eppendorf),Himac CR22GⅡ高速冷冻离冷冻离心机 (Hitachi),BIO-RAD荧光定量PCR仪iQ5 (Bio-Rad)。
1.1.6 引物
根据米根霉基因组序列数据库(http://www. broadinstitute.org/annotation/genome/rhizopus_oryzae/MultiHome.html) 中各个基因的搜寻结果,结合与NCBI数据库中的相关数据的比对分析,针对各个基因片段及部分启动子序列及终止子序列设计引物 (表2)。
1.2 方法
1.2.1 重组基因PA--TA及PA-A-TA的合成
从米根霉斜面上制取孢子悬液,调节孢子悬液终浓度为1×105个/mL,并按5% (/) 接种量接种到菌丝体培养基中。32 ℃振荡培养12 h左右。当培养液中有均匀分散的米根霉菌丝体时便可取出进行基因组DNA的提取。米根霉菌体基因组提取过程参照分子克隆实验指南有关内容并加以改进[14]。
按照表2中的说明,以基因组DNA为模板进行融合前的各个片段的扩增。片段回收并克隆后选取菌落PCR及酶切验证成功的转化子送上海生工生物技术服务有限公司测序,并进行同源性比对及分析。扩增片段的回收纯化、连接、转化大肠杆菌、质粒提取、酶切及回收等过程参考分子克隆实验指南[14]。
按照表2中各个引物设计以米根霉基因组DNA以及pBC-hygro质粒为模板,应用DNA聚合酶分别扩增合成重组基因所需的各个元件PA、、TA、PA以及A平末端片段,并回收纯化,以作为基因融合扩增的模板。
PATA基因融合参考文献中的方法并加以优化[13]。第一轮融合扩增分别用3种纯化后的PA和TA片段 (各1 μL) 作为扩增融合基因的模板,不加任何引物,以DNA聚合酶配置25 μL的PCR体系,按以下程序进行第一轮融合扩增:94 ℃ 3 min;94 ℃ 30 s,46.5 ℃ 45 s,72 ℃ 6 min,10个循环;72 ℃ 7 min。之后在第一轮扩增产物中加入hph_fu_F以及hph_fu_R引物各1.5 μL,并加入DNA聚合酶等,配置50 μL的PCR反应体系,进行第二轮融合扩增,程序如下:94 ℃ 3 min;94 ℃ 30 s,44.5 ℃ 45 s,72 ℃ 3 min,30个循环;72 ℃ 7 min。

表2 PCR所用引物及其序列
F and R in the primer names refer to upstream primer and downstream primer respectively; bases of restriction sites and protection bases are shown in lowercase letters;ⅠandⅠrestriction sites are underlined.
PAATA第一轮融合反应中的退火温度为43 ℃,第二轮融合扩增反应的退火温度为48 ℃,采用引物为ldhA_fu_F以及ldhA_fu_R。
1.2.2 整合表达载体pBS-hygro-A的构建
整合表达载体pBS-hygro-A的构建方法如图1所示。将PATA以及PAATA融合片段与载体pBluescript SK+分别通过Ⅰ以及Ⅰ限制性内切酶位点进行连接。重组载体构建中的酶切连接过程参照分子克隆实验指南[14]。
1.2.3 原生质体制备
参考邱静等[15]的方法制备米根霉原生质体。菌丝体洗涤及原生质体渗透剂采用0.6 mol/L山梨醇溶液。之后参照文献[15]的方法进行菌丝体酶解以及原生质体收集。
1.2.4 原生质体PEG/CaCl2介导转化
原生质体用含0.6 mol/L 山梨醇的Tris-HCl溶液 (pH 7.5,10 mmol/L) 洗涤,500 r/min、10 min离心2次,然后以同样的溶液重悬并镜检计数,调节原生质体悬液体积使原生质体浓度达到1×107个/mL以上。将5 μg (≤10 μL) 经过Ⅰ限制性内切酶线性化的整合载体与100 μL原生质体混合,加入25 μL的含25% (/) PEG 6 000、50 mmol/L CaCl2的Tris-HCl (pH 7.5,10 mmol/L) 溶液,冰浴20 min。之后再次加入1 mL上述Tris-HCl溶液,混匀后25 ℃放置10 min。加入5 mL的原生质体再生培养基,80 r/min轻摇再生预培养5 h。浓缩体积至150 μL用于涂板 筛选。

图1 表达载体pBS-hygro-ldhA构建示意图
1.2.5 原生质体电转化
原生质体用0.6 mol/L山梨醇溶液洗涤,用含0.6 mol/L山梨醇、5 mmol/L LiAc、10 mmol/L DTT的溶液重悬并室温放置1 h,之后以含 2 mmol/L HEPES (pH 7.5)、1 mmol/L LiAc、 0.6 mol/L山梨醇的溶液洗涤2次,清洗后的原生质体最终重悬于上述溶液中,镜检计数,调节悬液体积使原生质体浓度达到1×107个/mL以上。将5 μg (≤10 μL) 经过Ⅰ限制性内切酶线性化的整合载体与60 μL原生质体悬液混合均匀,冰浴20 min。使用0.1 cm电击杯中,于12 kV/cm的电场强度下电击5 ms对感受态细胞进行电转化处理。然后加入5 mL原生质体再生培养基,80 r/min轻摇再生预培养5 h,之后浓缩体积至150 μL用于涂板筛选。
1.2.6 萌发孢子制备及电转化
参考李松等[16]的方法进行米根霉萌发孢子电转化。接种米根霉孢子于菌丝体液体培养基中使孢子终浓度达到1×107个/mL以上,于32 ℃、200 r/min摇瓶培养3 h萌发。收集并洗涤后的萌发孢子用0.6 mol/L山梨醇溶液洗涤2次,4 000 r/min离心10 min收集,用含270 mmol/L蔗糖、5 mmol/L LiAc、10 mmol/L DTT的溶液重悬并室温放置1 h。之后以含2 mmol/L HEPES (pH 7.5)、1 mmol/L LiAc、270 mmol/L蔗糖的溶液洗涤2次,4 000 r/min离心10 min。清洗后的萌发孢子最终重悬于上述洗涤溶液中,镜检计数,调节悬液体积使萌发孢子浓度达到1×107个/mL以上。按照1.2.5方法进行电转化处理,电场强度为15 kV/cm。
1.2.7 转化子筛选
将原生质体及萌发孢子转化后的菌体悬液分别涂布于原生质体筛选培养基及筛选培养基平板上,同时以不加质粒的转化体系的菌体作为对照。32 ℃培养4−6 d直至抗性转化子菌落出现。挑选菌落点种于筛选培养基平板上进行培养验证其抗性稳定性,并以无菌蒸馏水洗脱收集产孢菌株的孢子,稀释后再次涂布于筛选培养基平板上进行二次验证。筛选出的具有稳定抗性的转化子提取基因组作为模板,以hph_F及hph_R为引物进行PCR验证,确定所筛选出的抗性转化子为阳性转化子。并且以每μg线性化重组质粒DNA得到的经过PCR验证的阳性转化子数计算各种转化方法的转化率。
1.2.8 原生质体再生率
1.2.4和1.2.5中分别经过转化处理后在预培养之前取出10 μL原生质体悬液加入990 μL的0.6 mol/L山梨醇溶液中,摇匀稀释后镜检计数。并分别取100 μL悬液涂布于PDA固体培养基和原生质体再生固体培养基平板上,32 ℃培养12 h后进行菌落计数,按照公式1进行再生率计算。

1.2.9 米根霉荧光定量PCR检测及转化子A基因拷贝数测定
选择米根霉AS 3.819的A基因作为对象,应用荧光定量PCR (qPCR) 技术对转化子基因组DNA进行检测。依照1.2.1中所述方法,从原始米根霉以及转化子菌株中提取基因组DNA。使用qPCR引物ldhA_qPCR_F和ldhA_qPCR_R,以基因组为模板,进行qPCR检测。同时,以米根霉AS 3.819的葡糖糖化酶A基因 (Glucoamylase A,A) 作为内参 (Internal control),以引物amyA_qPCR_F和amyA_qPCR_R对同等量的基因组中A基因进行qPCR检测,并以此结果对原始米根霉菌株对照及转化子菌株中A的qPCR检测结果进行归一化校正。

2 结果与分析
2.1 重组基因PamyA-hph-TpdcA及PpdcA- ldhA-TpdcA的合成
以1.5中介绍的方法扩增得到PA、、TA、PA以及A片段,其大小分别为899、1 026、805、1 083以及966 bp,各个片段的电泳检测结果如图2所示。
经过两次两轮的融合PCR反应,获得了两种融合片段PATA与PAATA,大小分别为2 724 bp和2 848 bp,与预期的大小相吻合,融合片段电泳检测如图3所示。融合片段送往上海生工生物技术服务有限公司测序,对比结果表明DNA片段融合顺序正确。由此可以判断,应用重叠延伸PCR的方法,正确地得到了两条分别由3段DNA片段以一定顺序融合而成的融合基因表达盒片段。
2.2 重组载体pBS-hygro-ldhA的构建
按照1.2.2所描述的方法构建载体,并转化大肠杆菌JM 109,经过筛选后提取得到的质粒经过对A及片段PCR验证及分别以Ⅰ和Ⅰ酶切验证正确后,送往上海生工生物技术服务有限公司以M13引物进行测序分析,测序对比结果表明两条融合片段正确地连接到了载体的Ⅰ和Ⅰ位点上,表达载体pBS-hygro-A构建成功。分析测序结果后,在测序鉴定连接上了融合片段的重组载体中,优先选择了PAATA与PATA 的启动方向相反的质粒,进行米根霉的转化 操作。
2.3 多拷贝表达ldhA基因的重组米根霉的构建
用测序鉴定连接成功并连接方向正确的质粒pBS-hygro-A对米根霉进行转化。携带潮霉素B抗性的数株转化子经过划线传代接种于抗性筛选平板上,部分菌株能稳定生长。挑选3株转化子提取基因组DNA为模板,以hph_F及hph_R为引物进行PCR验证,并以重组质粒及米根霉原始菌株基因组样品为对照,其中有两株得到了与基因对应的1 kb左右相同大小的条带 (图4)。通过潮霉素抗性筛选出的米根霉转化子中存在一定的假阳性的现象。抗性复筛以及PCR验证表明部分抗性转化子中质粒未整合入基因组。在3种不同转化方法的筛选结果中,假阳性的结果约占总转化子数目的20%−30%。

图2 ldhA、hph、PpdcA、PamyA以及TpdcA片段的扩增结果

图3 片段PamyA-hph-TpdcA与PpdcA-ldhA-TpdcA的融合PCR产物
2.4 原生质体转化条件研究
2.4.1 菌丝酶解时间对转化效果的影响
菌丝体沉淀加入菌丝酶解溶液后以不同时间进行酶解,制取的原生质体经感受态制备并调节浓度一致之后用于进行原生质体再生及转化实验。如图5所示,酶解时间为100–140 min时,制备的原生质体经PEG/CaCl2介导转化及电转化处理后其再生率都能保证在相对较高的水平 (最高在120 min分别达到10.2%和8.7%的再生率),当酶解时间小于100 min时,根据公式1所计算出来的再生率相反要比酶解100 min的情况下低。之后当酶解时间大于160 min原生质体再生率则出现锐减。原生质体转化率在 80 min至140 min时则逐渐升高,在120–140 min趋于平稳,当大于140 min则开始下降,大于160 min后转化率出现锐减。由此结果可以得知,无论对于PEG/CaCl2介导转化还是电转化方法,菌丝酶解120 min至140 min时间内所制得的原生质体都是适合于感受态细胞的制作,此时间段内制备的原生质体能够保持较高的活性,细胞壁去除完全,原生质膜也比较完整,有利于DNA的导入,其再生率也相较其他酶解时间产生的原生质体高。考虑原生质体获得数的增加可以减少收集原生质体的步骤从而减少操作过程中可能导致的原生质体的损失以及污染,并最大限度地节省时间,因此本研究中较优的菌丝酶解时间为140 min。
2.4.2 电场强度对原生质体电转化效果的影响
观察酶解2 h所制得的原生质体在不同电场强度下进行点击转化的再生及转化结果。如图6所示,随着脉冲电场强度的增加,转化后的原生质体细胞再生率逐渐下降。当电场强度达到13 kV/cm时,原生质体再生率由平稳开始下降,而在大于14 kV/cm后,原生质体再生率显著下降。这说明大于14 kV/cm的电场强度可能会对部分米根霉原生质膜造成不可逆的击穿,从而影响原生质体的存活再生。电场强度低于 13 kV/cm时,原生质体转化率随着电场强度的增加而提高,最高达到3.2个/μg DNA。当电场大于13 kV/cm时,转化率下降,此时的转化率更主要受到原生质体再生率下降的影响。在所选电场强度范围内,较优的原生质体电转化脉冲电场强度为13 kV/cm。

图4 PCR鉴定转化子米根霉基因组DNA
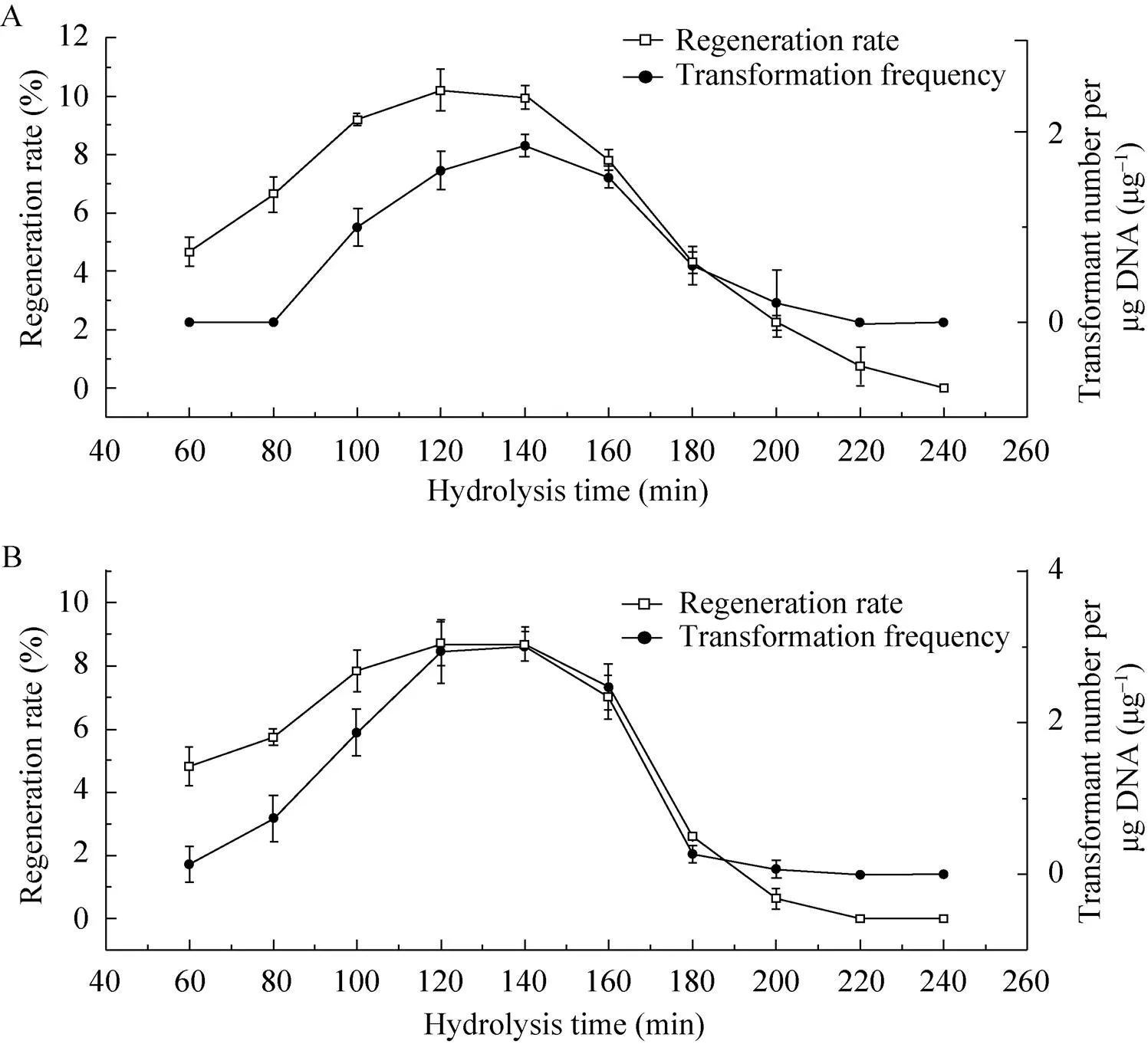
图5 菌丝酶解时间对原生质体再生率及转化率的影响

图6 电场强度对原生质体再生率及转化率的影响
2.5 萌发孢子电转化条件研究
2.5.1 萌发时间对转化效率的影响
分别取不同时间点的萌发孢子进行转化实验。在15 kV/cm电场脉冲条件下,转化结果如 图7A所示,萌发1 h时开始得到转化子,而后随着萌发时间的增加转化率提高,并在2.5 h时达到最高10.2个/μg DNA。之后转化率开始下降,在萌发4.5 h后萌发孢子无法被转化。镜检萌发孢子状态发现此时大部分孢子已经生成菌丝并已有部分聚集在一起,这可能是导致转化率急剧下降的主要原因之一。由此结果可知,初始未萌发的厚垣孢子由于壁厚抵抗能力强,无法通过电穿孔的方法将外源DNA导入孢子内部,同样萌发时间过长导致菌丝形成也不适合于用电穿孔的方式产生可逆孔洞,两种状态下都无法获得理想的电转化效果,而只有萌发一定时间,孢子厚壁消失而菌丝细胞壁未形成时,才可能通过电击形成孔洞使外源DNA进入。

图7 萌发时间和电场强度对萌发孢子转化率的影响
2.5.2 电场强度对转化效果的影响
观察萌发3 h的孢子在不同电场强度下的转化结果。与原生质体电转化的情况不同,如图7B所示,萌发孢子电转化所需电场强度整体要比原生质体电转化高,这可能是由于萌发孢子的外壁比原生质体膜要难以击穿,所以需要更高的脉冲电场才能达到同等的效果。在所选电场强度范围内,当达到14 kV/cm时,转化率达到最高8.8个/μg DNA。
2.6 添加Triton X-100及脱氧胆酸钠对筛选效果的影响
在含有20、50、80、100、120、150、180、200、250 μg/mL潮霉素B的筛选平板中分别各添加0.5 g/L浓度的脱氧胆酸钠及0.05% (/) 的Triton X-100,并分别涂布孢子悬液及预培养5 h的原生质体悬液。经过32 ℃培养观察菌落出现情况及菌落形态。同未加入Triton X-100和脱氧胆酸钠的筛选平板相比,潮霉素B对于菌体的抑制性皆有所提高。未添加Triton X-100和脱氧胆酸钠的情况下,米根霉对潮霉素B耐受上限浓度为200 μg/mL,含200 μg/mL以下浓度的筛选平板皆在4 d内长出米根霉菌落。而在添加了0.05%的Triton X-100之后米根霉对潮霉素B耐受浓度上限降低到了120 μg/mL,使得米根霉对于潮霉素B更加敏感。相较而言,0.5 g/L浓度的脱氧胆酸钠使耐受浓度上限降低到了50 μg/mL,对于米根霉潮霉素B敏感性的提高作用更强烈,但同时也可能说明了脱氧胆酸钠对米根霉生长的抑制性更强。
另一方面,观察到米根霉在添加0.05%的Triton X-100的筛选平板上长出的菌落形态和未添加的筛选平板上的菌落不同,呈点状分散,菌落直径较小,并未形成明显的菌丝蔓延现象,菌落之间可以清晰的分开。而添加0.5 g/L浓度的脱氧胆酸钠的筛选平板上长出的菌落和未添加的筛选平板上的菌落形态区别并不明显,仍然会有菌落扩大交织的现象,并不能起到有效限制菌丝蔓延的作用。
在丝状真菌菌株筛选中,通过在培养基中添加如DMSO、脱氧胆酸钠或Triton X等表面活性剂可以起到限制菌丝蔓延的作用,从而防止菌落之间出现混杂难分的现象,方便菌落的计数与挑选分离[17-18],在本研究中添加Triton X-100可以达到此效果。同时由于表面活性剂有增加细胞通透性的作用,使得抗生素对于细胞的作用更加明显,从而降低了抗生素耐受限度。因此添加适当浓度的表面活性剂对于生长顽强抗生素耐受能力较强或者对于抗生素耐受性不稳定的菌株可以起到一定的改善和稳定作用。
2.7 不同转化方法的转化效率及转化子稳定性
分别使用优化后的条件对米根霉进行转化实验,条件如下:
菌丝酶解时间为140 min,原生质体电转化电场强度为13 kV/cm,孢子萌发时间为2.5 h,萌发孢子电转化电场强度为14 kV/cm。
转化筛选后各种方法的转化率对比结果见图8A。结果显示,不同转化方法之间转化效率存在一定差别,相比较而言,利用萌发孢子,每μg重组质粒DNA所获得的抗性转化子数要明显高于利用原生质体。这可能是因为米根霉原生质体转化率受制于其相对较低的再生率。另外,PEG/CaCl2介导的原生质体转化相比原生质体电转化其转化率较低,虽然由2.4.1的结果可以说明PEG/CaCl2介导转化处理后的原生质体再生率比电转化要高,但其将DNA导入米根霉细胞内的能力却要差于电穿孔的方式。

图8 三种转化方法的转化效率及转化子稳定性
在不同方法获得的转化子中各选出40株,分别点种转接到不含潮霉素B的PDA固体培养基平板上传代培养,每代分别再转接到含有潮霉素的PDA固体筛选平板上,如此反复传代10代,检验传代后米根霉转化子菌株的抗生素抗性是否稳定。如图8B所示,3种转化方法所得到的转化子在传代多次之后均有部分菌株出现潮霉素B抗性丢失,在传代10代之后尚有22−28株的菌株仍保持潮霉素B抗性,且潮霉素B抗性的丢失多发生在第2−5代。结合2.3的结果可知,采用本研究构建的pBS-hygro-A整合型表达载体转化米根霉AS 3.819菌株所获得的拥有稳定抗性的米根霉转化子占初筛获得的所有转化子的40%−50%左右。
2.8 转化子稳定性与质粒拷贝数的关系
应用qPCR的方法对转化子的质粒整合拷贝数进行了检测。从2.7部分所考察的转化子当中挑选菌株进行检测,在不同传代代数丢失抗性的转化子菌株中挑选10株,记为unstable #1−10;具有稳定抗性的转化子中挑选20株,记为stable #1−20。转化子qPCR结果见表3,这30株转化子的基因相对整合拷贝数在2到51范围内。从总体来说拥有相对较高质粒拷贝数也就是的拷贝数的转化子抗性相对较为稳定,但这种关系也不是严格对应的。在unstable #1−10中有两株相对stable #1−20拥有较高的拷贝数,却在传代中丢失抗性,而stable #1−20中也有部分拥有较低拷贝数。对于应用整合型质粒的遗传转化筛选而言,质粒拷贝数影响着质粒内基因 (包括抗性标记基因和目的基因) 的表达水平,从而一定程度上影响着宿主对于抗生素的耐受性,当达到拷贝数的阈值后,宿主就可以在一定浓度的抗生素筛选平板上生长。在本实验中,这种拷贝数的阈值表现得并不是非常的清晰明显,在一定拷贝数范围内,米根霉宿主抗性的稳定性会出现波动,更高的拷贝数可能会更有利于米根霉转化子的稳定性。然而质粒拷贝数过高会对宿主的生长造成一定影响,从而在传代当中菌株生长衰退滞后,最终无法再在抗性平板上生长。
3 讨论
基于遗传转化操作发展的代谢工程技术是研究代谢产物合成途径,调节其产量的重要技术之一在丝状真菌当中,遗传转化技术已得到一定的应用,部分丝状真菌如脉孢霉属的粗糙脉孢霉,曲霉属的构巢曲霉、米曲霉、黑曲霉以及木霉属的瑞氏木霉、绿色木霉等已经建立了相对成熟可靠的遗传转化体系及方法。目前,包括米根霉在内的部分丝状真菌其基因转化筛选方法是以营养缺陷型标记为基础建立的[19-20]。如米根霉采用脲嘧啶营养缺陷型标记为筛选标记,宿主米根霉需要诱变使脲嘧啶合成酶系发生缺陷,以与载体的脲嘧啶合成相关酶基因G或F形成互补,达到筛选目的[6,8,10]。脲嘧啶合成酶系缺陷菌株是特定酶基因突变导致的营养缺陷型菌株,其适用范围受到一定的限制,使用抗生素抗性作为筛选标记在这种情况下便显现出了一定的优势。随着各种稳定有效的抗生素抗性筛选标记在丝状真菌遗传转化中的成功应用,简便而稳定的转化体系的建立也已成为了丝状真菌遗传转化研究的重要内容。选择合适的筛选标记及DNA转化方法对米根霉遗传转化体系的建立是十分重要的。在本研究中,我们尝试使用潮霉素B抗性作为筛选标记,对米根霉的抗生素耐受性进行了先行实验,得到了米根霉AS 3.819菌株对潮霉素B的最高耐受浓度 (200 μg/mL),并且针对米根霉自身菌落生长情况进行了改良,使得潮霉素B抗性筛选操作更适宜于米根霉菌株。除此之外,我们在前期研究中还试验了米根霉对G418 (即遗传霉素Geneticin),博莱霉素及新霉素的耐受性,其中米根霉对于较高浓度的博莱霉素 (>500 μg/mL) 仍然有着一定的耐受性,而对于新霉素其最大耐受浓度常常显现出不稳定的现象,不同平行实验下得到的最大耐受浓度常常显现出较大的波动。此外,米根霉AS 3.819对于平常适用于大部分酵母及其他部分真菌的G418却能够表现出一定的敏感性,250 μg/mL浓度的G418可以完全抑制米根霉AS 3.819菌株的生长。因此,基于G418抗性筛选标记的转化筛选方法也有应用于米根霉遗传操作研究当中的可能。

表3 米根霉转化子基因整合拷贝数
Tresults are presented as average values of 3 parallel experiments.
应用于丝状真菌遗传转化研究的常用转化方法主要包括原生质体PEG介导化学转化、原生质体LiAc介导电穿孔转化、基因枪转化及农杆菌介导转化。其中使用原生质体为材料的转化方法发展较早,应用相对比较广泛,然而使用原生质体作为材料进行转化其转化效率十分依赖于菌株原生质体的制取效率和再生率,对酶解时间及原生质体收集、转化操作的条件如果没有详实的了解及严谨的控制,则会导致DNA转化效率大大降低甚至很难得到转化子。本研究针对米根霉AS 3.819研究了适用于其遗传操作的原生质体制备和转化条件,得到了拥有潮霉素抗性的转化子。但在本研究的转化操作条件下,原生质体再生率只有7%−10%左右,由于受到再生率的影响,转化率也相对较低。因此原生质体转化的手段如要很好地应用于米根霉的遗传转化研究中,提高原生质体获取产量和质量,以及再生率将是需要考虑的重要问题之一。除此之外,也可以选择应用其他转化效率相对较高的转化方法,如已应用于米根霉的基因枪转化方法[6-10],本研究所使用的萌发孢子电转化方法,以及农杆菌介导转化[5]等方法。其中基因枪转化方法和农杆菌介导的转化方法在丝状真菌当中得到了越来越广泛的应用,由于其转化目标菌株无需特殊处理,适用微生物及组织范围广,并且其转化效率及质粒整合入基因组的稳定性也高于其他转化方法,因此也得到越来越多的研究者的肯定。本研究所使用的萌发孢子电转化方法同样也已应用于诸如马尔尼菲青酶[21]、小单孢菌[22]及黑曲霉[16]等的遗传转化研究当中,相比基于原生质体的转化方法,由于不需要考虑再生过程造成的损失,其操作难度和转化效率方面都具有一定的优势,适用于能够产生一定数量孢子,或者较难制备原生质体的丝状真菌。
本研究选择潮霉素B抗性为筛选标记,构建了整合型表达载体pBS-hygro-A,以A为单交换重组同源臂在米根霉AS 3.819基因组中进行多拷贝整合,成功获取了重组菌株,建立了区别于营养缺陷型筛选的另一套适用于普通工业应用的米根霉菌株的遗传转化筛选方法。依此研究希望为米根霉代谢工程改造及工业应用价值扩展提供帮助。
[1] Bai DM, Zhao XM, Li XG, et al. Strain improvement offor over-production of L(+)-lactic acid and metabolic flux analysis of mutantsBiochem Eng J, 2004, 18(1): 41–48.
[2] Jiang ST, Zheng Z, Zhu Y, et al. Repeated intermittent L-lactic acid fermentation technology by self-immobilizedChin J Biotech, 2008, 24(10): 1729–1733 (in Chinese).姜绍通, 郑志, 朱羽, 等. 无载体固定化米根霉重复间歇发酵生产L-乳酸. 生物工程学报, 2008, 24(10): 1729–1733.
[3] Zhou Q, Zhao L, Jiang XW, et al. Astrain producing L-lactic acidMycosystema, 2012, 31(6): 956–962 (in Chinese).周倩, 赵龙, 蒋雪薇, 等. 一株产生L-乳酸的米根霉. 菌物学报, 2012, 31(6): 956–962.
[4] Ma LJ, Ibrahim AS, Skory C, et al. Genomic analysis of the basal lineage fungusreveals a whole-genome duplicationPLoS Genet, 2009, 5(7): e1000549.
[5] Michielse CB, Salim K, Ragas P, et al. Development of a system for integrative and stable transformation of the zygomyceteby-mediated DNA transferMol Genet Genomics, 2004, 271(4): 499–510.
[6] Skory CD. Homologous recombination and double-strand break repair in the transformation ofMol Genet Genomics, 2002, 268(3): 397–406.
[7] Skory CD. Inhibition of Non-Homologous end joining and integration of DNA upon transformation ofMol Gen Genomics, 2005, 274(4): 373–383.
[8] Mertens JA, Skory CD, Ibrahim AS. Plasmids for expression of heterologous proteins inArch Microbiol, 2006, 186(1): 41–50.
[9] Skory CD, Ibrahim AS. Native and modified lactate dehydrogenase expression in a fumaric acid producing isolate99-880Curr Genet, 2007, 52(1): 23–33.
[10] Zhang BH, Skory CD, Yang ST. Metabolic engineering of: effects of overexpressingandgenes on fumaric acid biosynthesis from glucoseMetab Eng, 2012, 14(5): 512–520.
[11] Robert CD, Jill RB, Peter RK, et al. A PCR-based strategy to generate integrative targeting alleles with large regions of homologyMicrobiology, 2002, 148(8): 2607–2615.
[12] Majid Z, Abigail CL, Geoffrey T. A rapid method for promoter exchange inusing recombinant PCRFungal Genet Biol, 2005, 42(1): 1–8.
[13] Charlier N, Molenkamp R, Leyssen P, et al. A rapid and convenient variant of fusion-PCR to construct chimericJ Virol Methods, 2003, 108(1): 67–74.
[14] Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 3rd ed. New York: Cold Spring Harbor Laboratory Press, 2001.
[15] Qiu J, Luo SZ, Jiang ST, et al. Preparation and regeneration of high-yield L-lactic acid-producing protoplast fromFood Sci, 2011, 32(9): 174–178 (in Chinese).邱静,罗水忠,姜绍通, 等. 高产L-乳酸水根霉的原生质体制备与再生条件研究. 食品科学, 2011, 32(9): 174–178.
[16] Li S, Wang ZX. Transformation ofby electroporation-mediated transformationJ Microbiol, 2010, 30(1): 11–15 (in Chinese).李松, 王正祥. 黑曲霉电转化条件的研究. 微生物学杂志, 2010, 30(1): 11–15.
[17] Notman R, Noro M, O'Malley B, et al. Molecular basis for dimethylsulfoxide (DMSO) action on lipid membranesJ Am Chem Soc, 2006, 128(43): 13982–13983.
[18] Wang YL, Hu PJ, Li EW, et al. Genetic transformation of the fungussp. mediated byActa Microbiol Sin, 2013, 53(11): 1233–1239 (in Chinese).王艳玲, 胡鹏杰, 李二伟, 等. 农杆菌介导的粘帚菌遗传转化. 微生物学报, 2013, 53(11): 1233–1239.
[19] Ji YW, Xu Y, Li YP, et al. Application of membrane filtration method to isolate uninuclei conidium intransformation system based on theG markerFood Sci Biotechnol, 2013, 22(1): 93–97.
[20] Du Y, Xie GZ, Yang CF, et al. Construction of brewing-wineG(-) mutant byG gene deletion and its application in homology transformationActa Bioch Bioph Sin, 2014, 46(6): 477–483.
[21] Chen YX, Gu AJ, Liang YH, et al. Electroporation-mediate transformation ofand optimization of the transforming conditionsChin J Mycol, 2014, 9(1): 12–15 (in Chinese).陈阳霞, 顾桉菁, 梁宇恒, 等. 电穿孔介导马尔尼菲青霉转化体系的建立和优化. 中国真菌学杂志, 2014, 9(1): 12–15.
[22] Li XH, Long CF, Zhou XF, et al. Study on electroporation of raresp. 40027 withplasmid pSET152Acta Microbiol Sin, 2007, 47(4): 718–720 (in Chinese). 李晓华, 龙慈凡, 周秀芬, 等. 链霉菌质粒pSET152电转化稀有放线菌小单孢菌的研究. 微生物学报, 2007, 47(4): 718–720.
(本文责编 郝丽芳)
Construction of an integration vector carrying hygromycin B resistance gene and its genetic transformation in
Min Zhang, Shaotong Jiang, Juan Zheng, Zhi Zheng, Xingjiang Li, Lijun Pan, and Shuizhong Luo
,,230009,,
To construct a system of genetic transformation suitable for, we constructed a single-exchange vector pBS-hygro carrying hygromycin B resistance gene () as its selective marker using gene splicing by overlap extension PCR (SOE PCR) technique. We introduced this recombinant vector intoAS 3.819 by PEG/CaCl2-mediated transformation of protoplast, electroporation of protoplast and germinated spores; and we studied the effects of hydrolysis time, field strength and spore germination time on transformation frequency. We conducted quantitative real-time PCR (qPCR) assay to determine the gene copy number ofA integrated in the genome of.transformants and its effect on the stability of transformants. We successfully achieved.transformants integrated with pBS-hygro-A vector. The optimal hydrolysis time for protoplast production was 140 min, and the optimal field strength of electroporation pulse for protoplast was 13 kV/cm. The optimal germination time of spores for electroporation was 2.5 h, and the optimal field strength of electroporation pulse was 14 kV/cm. The transformation frequency of method based on germinated spores was generally higher than the methods based on protoplast. The qPCR test results suggested that transformants with high copy number of integration in a certain range were relatively stable. Our results provided basis and support for metabolic regulation and genetic engineering breeding of..
, integration vector, protoplast, germinated spore, homologous recombination, gene splicing by overlap extension PCR, electroporation
10.13345/j.cjb.140490
October 20, 2014; Accepted: January 4, 2015
National Natural Science Foundation of China (Nos. 31071636, 31171741, 31101352, 31470002).
Shaotong Jiang. Tel: +86-551-2901505-8316; Fax: +86-551-2901507; E-mail: zming19861028@hotmail.com
国家自然科学基金 (Nos. 31071636, 31171741, 31101352, 31470002) 资助。
2015-02-03
http://www.cnki.net/kcms/detail/11.1998.q.20150203.1614.002.html
猜你喜欢
种子(2023年4期)2023-06-19 01:15:08
河北医学(2021年10期)2021-10-27 00:37:14
江苏农业科学(2021年1期)2021-03-15 06:06:41
甘肃农业科技(2019年8期)2019-09-10 07:22:44
中国临床医学影像杂志(2019年6期)2019-08-27 02:59:50
食用菌(2017年5期)2017-10-19 03:02:28
中国科技博览(2016年24期)2016-12-28 17:44:12
大连工业大学学报(2015年4期)2015-12-11 04:06:50
特产研究(2015年1期)2015-04-12 06:36:11
发明与创新(2015年25期)2015-02-27 10:39:16
