
多发性内分泌腺瘤病1型一例
2015-07-18 11:24郭宜晨段山杨川
新医学 2015年9期
郭宜晨 段山 杨川
综合病例报告
多发性内分泌腺瘤病1型一例
郭宜晨 段山 杨川
多发性内分泌腺瘤病(MEN)1型是一种常染色体显性遗传的内分泌肿瘤综合征,是由MEN1抑癌基因的失活突变引起的,主要临床表现有甲状旁腺功能亢进、胰腺内分泌肿瘤、垂体腺瘤等。其诊断主要依靠临床资料及综合检查结果。近年来基因检测技术不断发展,基因诊断在提高MEN1型的诊断率中发挥了重要作用,然而MEN1型患者的临床症状及病情轻重各有其自身特点,基因诊断在临床工作中的应用仍存在争议。该文总结1例以皮质醇增多症(库欣综合征)起病,经基因检测发现MEN1基因第10外显子存在新的杂合突变的MEN1型患者的临床资料、治疗及随访情况,并复习了相关文献。综合分析临床资料及各项检查结果、合理运用基因检测、个体化治疗方案及有效的随访对改善患者预后和生活质量有重要临床意义。
多发性内分泌腺瘤病1型;基因突变;个体化治疗;随访
多发性内分泌腺瘤病(MEN)是指患者身上同时或相继发生2种或2种以上内分泌腺增生或肿瘤,呈常染色体显性遗传,临床上主要分MEN1型和MEN2型,后者又分为MEN2A、MEN2B亚型。其中MEN1型的发病率为1/3万~1/5万,外显率较高,常见的临床表现有甲状旁腺腺瘤、胃胰肠肿瘤和垂体瘤。患者有上述3 种内分泌肿瘤中的2种即可诊断为MEN1型, 若有至少1个1级亲属也患有上述3 种内分泌肿瘤中的1种则可诊断为家族性MEN1型,若缺乏家族史则可诊断为散发的MEN1型[1]。本文报道1例以皮质醇增多症(库欣综合征)起病的MEN1型患者的临床特点及其诊治过程,并结合文献进行讨论。
病例资料
一、病史及体格检查
患者女,41岁,因视物模糊、体型改变2月余于2013年6月入我院。患者2月余前无明显诱因出现视物模糊、双眼复视,并逐渐出现面部肥大变圆、颈项增粗、腹部膨隆、四肢变细等体型变化,伴头痛、活动后乏力,于外院检查测得血压最高达140/100 mm Hg(1 mm Hg=0.133 kPa),空腹静脉血糖11.1 mmol/L,未做详细检查,且未接受规律治疗。患者既往无外用、口服、静脉使用糖皮质激素药物史。为进一步诊治而来我院就诊。入院时体格检查:血压 130/90 mm Hg,满月脸,双眼结膜充血,水牛背、向心性肥胖,全身皮肤菲薄;腹部膨隆,未见皮肤紫纹,四肢相对较细。其余体格检查未见明显异常。
二、实验室及辅助检查
1.实验室检查
血皮质醇及ACTH水平显著升高、昼夜节律消失;24 h尿游离皮质醇(24 h UFC)显著升高;小剂量地塞米松抑制试验(LDDST)不受抑制,大剂量地塞米松抑制试验(HDDST)可受抑制。血甲状旁腺激素(PTH)水平显著升高,多次测定血钙在正常上限或轻度升高,血磷、碱性磷酸酶(ALP)、尿钙均在正常范围;血胃泌素水平显著升高。其他内分泌检查:甲状腺功能正常,甲状腺过氧化酶抗体、甲状腺球蛋白抗体正常;血雌二醇、卵泡刺激素(FSH)、黄体生成素(LH)、孕酮符合黄体期水平,泌乳素、睾酮、生长激素均在正常范围;血儿茶酚胺、肾素活性、醛固酮均在正常范围,见表1 。
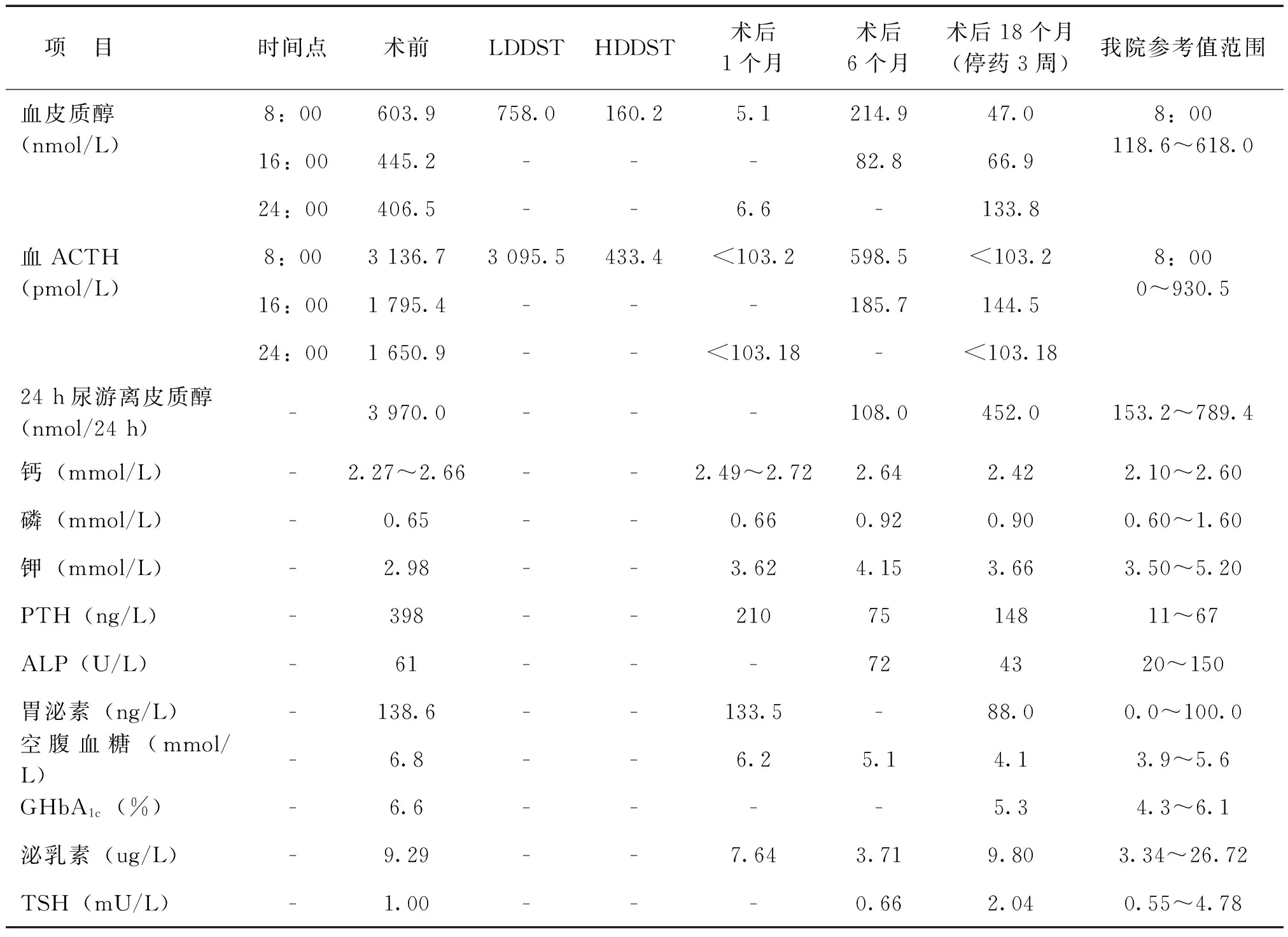
表1 本例MEN1型患者实验室检查结果
2.辅助检查
内镜检查:胃镜示慢性浅表性胃窦炎伴糜烂。全身X线片未见骨质破坏表现。甲状腺超声:甲状腺未见明显占位。腹部超声未见占位性病变。垂体MRI:垂体前叶结节(大小约9 mm×7 mm×3.2 mm),考虑垂体腺瘤(图1)。肾上腺MRI:双侧肾上腺增生,左侧肾上腺结节样增生。腹部及盆腔MRI:双侧卵巢囊肿。甲状旁腺同位素显像:甲状腺右叶下方99mTc-MIBI显像阳性(剂量25 mCi),考虑甲状旁腺瘤可能。
患者存在2种以上内分泌腺体肿瘤,临床上高度怀疑MEN1型可能,因此我们对该患者进行了基因检测。提取患者外周血白细胞DNA进行MEN1基因的9个编码外显子的PCR扩增及直接测序,引物设计依据文献[2],经过与GenBank数据库标准序列(NG_008929.1)对比共发现2处变异,分析后考虑在第9外显子处的变异为无意义的突变(rs2071313),而在第10外显子发现的杂合突变(C.1731 G>A,P.541A>T)使编码序列1 731位碱基G突变为A,从而使541位丙氨酸突变为苏氨酸,该突变在国人中未曾发现,经PubMed行文献搜索发现该突变为一新的突变类型[3-7]。采用工具Phyre2对该突变导致所翻译的蛋白质结构进行预测,蛋白质三级结构未见明显改变,但其生物学功能是否有变化则需要进一步行相关实验证实。
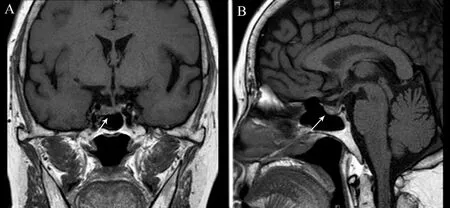
图1 本例MEN1型患者垂体MRI图
A:冠状面扫描可见垂体前叶异常信号结节,垂体柄向左侧轻度偏斜(箭头);B:矢状面扫描可见垂体前叶内类椭圆形异常信号结节(箭头),垂体上缘膨隆,视交叉无明显受压,考虑垂体腺瘤
三、诊断及治疗
患者于2013年6月接受经鼻蝶入路垂体瘤显微切除术治疗,术后病理检查示:垂体腺瘤,CK(+)、Vimentin(+)、Syn(+)、CgA(+)、ACTH(+)、NSE部分(+)、Ki67约3%(+),见图2。该患者垂体腺瘤ACTH瘤、甲状旁腺功能亢进症、胃泌素分泌增多诊断明确,存在MEN1型中3个主要病变,且在MEN1基因编码区第10个外显子发现新型杂合突变。因其一级亲属未发现上述肿瘤病变,综合临床表现、病理检查及基因检测结果,临床诊断为散发的MEN1型。行垂体瘤切除术后,患者出现肾上腺皮质功能减退,予醋酸泼尼松片口服替代治疗后症状好转;针对甲状旁腺功能亢进方面,患者拒绝接受手术治疗,遂要求其定期返院复查PTH、血钙及血磷水平,但仍建议患者择期行手术切除,同时定期复查胃泌素。

图2 本例MEN1型患者垂体腺瘤病理检查图(苏木素-伊红染色)
A:可见垂体腺瘤肿瘤细胞呈条索状排列(×100);B:高倍镜视野下可见胞浆呈嗜酸性染色,细胞核大小一致,分布均匀(×400)
四、随访及转归
对此例患者我们制定了持续随访计划,每半年复查血皮质醇、ACTH水平及节律、血清PTH、钙、磷、胃泌素及其他重要生化指标,每1年复查甲状旁腺超声、垂体MRI、腹部MRI等影像学检查。术后6个月(2013年12月)随访患者,库欣综合征面容消失,临床症状明显改善,肾上腺皮质功能仍低下,在使用生理剂量醋酸泼尼松替代治疗的情况下,其血皮质醇、ACTH水平在正常范围,PTH较前下降,甲状腺轴、性腺轴激素水平正常,血糖、电解质及其他激素水平均在正常范围。随访至术后18个月(2014年12月),患者的肾上腺皮质功能仍低下,继续予生理剂量醋酸泼尼松替代治疗,术后18个月的检查结果较术后6个月的检查结果无明显变化。截至撰稿前(2015年2月)患者病情稳定。
讨 论
本例患者有如下特点: ①中年女性,以典型库欣综合征症状起病,血皮质醇水平明显升高,呈ACTH依赖性,垂体影像学检查发现微腺瘤(<10 mm),术后病理检查结果为垂体ACTH瘤;②血液生化检验提示甲状旁腺功能亢进,血PTH水平明显升高,血钙轻度升高或在正常上限,甲状旁腺同位素显像提示甲状腺右叶下方99mTc-MIBI显像阳性,血PTH水平在行垂体瘤切除术后下降;③胃泌素分泌增多,无难治性胃溃疡,影像学未发现占位性病变;④外周血白细胞DNA测序发现,在MEN1基因第10个外显子发现杂合突变,此突变导致541位丙氨酸突变为苏氨酸,文献检索证实该突变为新的突变类型;⑤垂体瘤切除术后出现肾上腺皮质功能低下,术后6、18个月复查,临床症状及生化指标较术前明显改善,但肾上腺皮质功能低下未能恢复,需长期接受激素替代治疗。
MEN1型是在同一患者身上同时或先后出现2种或2种以上的内分泌腺体肿瘤或增生的一种临床综合征。本病虽为罕见疾病,但其基因突变携带者的外显率很高,其复杂的临床表现容易导致漏诊。此类患者有其自身的临床特点,通过总结本例患者的诊治经验,我们认为在MEN1型的诊治过程中应注意以下几点。
早期发现非常重要。由于MEN1型的多种临床表现既可同时出现,也可先后出现,2种腺体病变的出现可相距长达20年,而与MEN1型相关的各种内分泌肿瘤有潜在恶性并更具侵袭性,其治疗原则亦有所不同,鉴别某单一内分泌肿瘤是 MEN1型的早期表现还是单纯性内分泌肿瘤,对早期发现并确定相应的诊治计划尤为重要[8-10]。本例患者以库欣综合征起病而就诊,在入院后逐步完善检查发现垂体ACTH瘤及其它内分泌肿瘤。既往报道显示库欣综合征在MEN1型患者中较少见且出现较晚,但对于以库欣综合征表现起病的患者,作病因诊断时也应及时考虑到MEN1型的可能性,进行有针对性的筛查,这对于提高MEN1型的确诊率十分有意义[11]。
MEN1型患者可能有其临床特殊性。以本文患者为例,我们发现手术切除垂体瘤后其临床症状及生化指标显著改善,但术后出现肾上腺皮质功能低下,在术后18个月时仍未恢复,需接受长期激素替代治疗,而单纯ACTH微腺瘤患者术后较少出现这种情况[12]。其次,患者血PTH明显升高,但与甲状旁腺功能亢进有关临床症状较轻微,无胃肠症状、多饮多尿、骨骼改变、泌尿系结石等表现,而胃泌素升高水平不到正常上限的5~10倍,与文献报道一致[13-14]。值得注意的是,患者血钙水平仅偶有轻度升高,与血PTH升高水平并不一致,且在行垂体瘤切除术后,血PTH水平出现显著下降。考虑其原因可能有:①ACTH瘤导致体内皮质醇水平明显升高,大量皮质醇一方面可直接刺激PTH分泌、腺体增生,造成低磷酸盐血症,加快骨质吸收,另一方面皮质醇可抑制肠道钙吸收、抑制肾小管重吸收钙与磷,从而导致血钙相对降低[15];②目前学者们认为MEN1型的发病有遗传背景,然而对于MEN1型各腺体肿瘤的组织学特异性仍属未知,这些特殊表现可能与肿瘤自身的生物学特点有关,其病理生理机制仍有待探索。
合理把握基因检测在诊断中的价值。MEN1型的发病在根本上是由MEN1基因的失活突变引起的,MEN1基因是一个抑癌基因,定位于染色体11q13,编码610 个氨基酸的Menin蛋白[6, 16]。自从1997年MEN1基因被首次发现以来,超过500种MEN1基因突变已被报道,而这些突变多数会导致Menin蛋白结构改变及功能异常[4-5]。随着基因检测技术的不断发展,基因诊断在提高MEN1型的确诊率中发挥了重要作用。我们对本例患者进行基因检测,确实发现在MEN1基因编码区第10外显子存在杂合突变,此突变使541位丙氨酸突变为苏氨酸,经PubMed文献搜索证实其为一新的突变类型,进一步预测蛋白质三级结构,未发现明确的结构变化,因此该突变是否真正导致Menin蛋白生物学功能异常仍需作进一步实验证实。国内外现有研究暂未明确MEN1型的突变热点基因位点,而其基因型与表现型之间的联系亦尚未建立[6, 17]。MEN1型的定义核心为临床综合征,因而我们认为在MEN1型的诊断过程中应综合分析各项临床证据,对临床高度可疑的患者进一步行基因检测,可提高诊断的可信度,但考虑到基因的多态性及变异的复杂性,发现突变不能作为最终诊断的金标准,亦不建议广泛应用于对MEN1型患者的亲属筛查。
长期随访对MEN1型患者有重要意义。既往的研究观察显示,甲状旁腺功能亢进可在其他MEN相关的腺体病变出现0.2~20年后才得到诊断[9]。胃肠胰内分泌瘤有潜在恶性,约1/3 MEN1型患者的死亡是由此类肿瘤引起,但此类肿瘤多数体积较小且所处部位相对较深,定位诊断相对困难。因此,对疑诊MEN1型患者,应长期认真随访其重要生化指标。本例患者血PTH及胃泌素水平升高,虽然目前相关的临床表现轻微,但应加强复诊随访,警惕病情进展所致的不良后果。
综上所述,我们对MEN1型应给予足够重视,争取早期发现,结合临床特点及基因检测结果作出合理诊断,通过多学科合作诊治并对患者进行有效的长期随访,对降低本病的漏诊率及病死率尤为重要。MEN1型患者有其自身的临床特征,临床上应提高甄辨能力。
[1]廖二元. 内分泌学. 2版. 北京:人民卫生出版社,2007:1251.
[2]Xu L, Li X, Feng B, Ni Y, Wang H, Wang L. A novel mutation of the MEN1 gene in a Chinese kindred with multiple endocrine neoplasia type 1. Endocr J,2010,57(9):839-845.
[3]Dilley WG, Kalyanaraman S, Verma S, Cobb JP, Lara-mie JM, Lairmore TC. Global gene expression in neuroendocrine tumors from patients with the MEN1 syndrome. Molecular Cancer,2005,4(1):9.
[4]Kihara M, Miyauchi A, Ito Y, Yoshida H, Miya A, Kobayashi K, Takamura Y, Fukushima M, Inoue H, Higashiyama T, Tomoda C. MEN1 gene analysis in patients with primary hyperparathyroidism: 10-year experience of a single institution for thyroid and parathyroid care in Japan. Endocr J,2009,56(5):649-656.
[5]Lemos MC, Thakker RV. Multiple endocrine neoplasia type 1 (MEN1): analysis of 1336 mutations reported in the first decade following identification of the gene. Human Mutation,2008,29(1):22-32.
[6]Chung YJ, Hwang S, Jeong JJ, Song SY, Kim SH, Rhee Y. Genetic and epigenetic analysis in Korean patients with multiple endocrine neoplasia type 1. Endocrinology and Metabolism,2014,29(3):270.
[7]Liu W, Han X, Hu Z, Zhang X, Chen Y, Zhao Y, Ji L. A novel germline mutation of the MEN1 gene caused multiple endocrine neoplasia type 1 in a Chinese young man and 1 year follow-up. Eur Rev Med Pharmacol Sci,2013,17(22):3111-3116.
[8]李小英. 多发性内分泌腺瘤病. 中国实用内科杂志,2006,26(22):1763-1766.
[9]Gaztambide S, Vazquez F, Castano L. Diagnosis and treatment of multiple endocrine neoplasia type 1(MEN1). Minerva Endocrinol,2013,38(1):17-28.
[10]李国夫,王宁. 多发性内分泌腺瘤1型及其相关的垂体腺瘤的研究. 中国微侵袭神经外科杂志,2012,17(5):236-238.
[11]Alzahrani AS, Al-Khaldi N, Shi Y, Al-Rijjal RA, Zou M, Baitei EY, Amin T. Diagnosis by serendipity: Cushing syndrome attributable to cortisol-producing adrenal adenoma as the initial manifestation of multiple endocrine neoplasia type 1 due to a rare splicing site MEN1 gene mutation. Endocr Pract,2008,14(5):595-602.
[12]胡增春, 魏明海, 尹剑, 张波, 马辉. 265例垂体瘤术后并发症诊治分析. 医学与哲学,2015,36(B1):45-48.
[13]贾翼,杨兴无. 多发性内分泌瘤病的研究进展. 中华内分泌外科杂志,2011,5(4):277-279.
[14]Sakurai A, Suzuki S, Kosugi S, Okamoto T, Uchino S, Miya A, Imai T, Kaji H, Komoto I, Miura D, Yamada M, Uruno T, Horiuchi K, Miyauchi A, Imamura M, Fukushima T, Hanazaki K, Hirakawa S, Igarashi T, Iwatani T, Kammori M, Katabami T, Katai M, Kikumori T, Kiribayashi K, Koizumi S, Midorikawa S, Miyabe R, Munekage T, Ozawa A, Shimizu K, Sugitani I, Takeyama H, Yamazaki M. Multiple endocrine neoplasia type 1 in Japan: establishment and analysis of a multicentre database. Clin Endocrinol (Oxf),2012,76(4):533-539.
[15]廖二元. 内分泌与代谢病学. 3版. 北京:人民卫生出版社,2012:532.
[16]Fontaniere S, Duvillie B, Scharfmann R, Carreira C, Wang ZQ, Zhang CX. Tumour suppressor menin is essential for development of the pancreatic endocrine cells. J Endocrinol,2008,199(2):287-298.
[17]Shimazu S, Nagamura Y, Yaguchi H, Ohkura N, Tsukada T. Correlation of mutant menin stability with clinical expression of multiple endocrine neoplasia type 1 and its incomplete forms. Cancer Science,2011,102(11):2097-2102.
Multipleendocrineneoplasiatype1:onecasereport
GuoYichen,DuanShan,YangChuan.
DepartmentofEndocrinology,SunYat-senMemorialHospitalofSunYat-senUniversity,Guangzhou510120,China
,YangChuan
Multiple endocrine neoplasia type 1 (MEN1) is an autosomal dominant endocrine tumor syndrome caused by the inactivation and mutation of tumor suppressor gene MEN1. MEN1 encompasses clinical characteristics featuring hyperparathyroidism, pancreatic neuroendocrine tumors and pituitary adenomas, etc. The diagnosis of MEN1 mainly relies on clinical data and comprehensive examination outcomes. Along with recent development of gene detection technique, genetic diagnosis plays a pivotal role in enhancing the diagnosis rate of MEN1. However, MEN1 patients have diverse clinical manifestations and severity of diseases, application of gene detection technique in clinical practice remains controversial. Herein, we reported the clinical data, treatment and prognosis of one case of MEN1. The patient first presented with hypercortisolism (Cushing’s syndrome) as onset symptom. Gene detection identified a novel heterozygote G to A transition in exon 10. We also conducted relevant literature review. Comprehensive analysis of clinical data and examination outcomes, proper application of gene detection, individualized therapy and effective follow-up are of clinical significance to improve MEN1 patients’ prognosis and quality of life.
Multiple endocrine neoplasia type 1; Gene mutation; Individualized therapy; Follow-up
10.3969/g.issn.0253-9802.2015.09.016
510120 广州,中山大学孙逸仙纪念医院内分泌科(郭宜晨,杨川);518048 深圳,深圳市人口与计划生育科学研究所(段山)
,杨川
2015-03-11) (本文编辑:洪悦民)
猜你喜欢
中国急救医学(2022年2期)2022-11-15
心电与循环(2021年4期)2021-11-29
昆明医科大学学报(2020年12期)2021-01-26
健康之家(2020年7期)2020-11-02
疯狂英语·新读写(2020年3期)2020-06-06
中国医学影像技术(2020年12期)2020-01-14
中学数学杂志(2019年9期)2019-05-29
中国体育教练员(2017年2期)2017-07-31
中国眼镜科技杂志(2016年17期)2016-10-24
磁共振成像(2015年9期)2015-12-26
