
改性HBeta分子筛催化二甲醚转化制六甲基苯的研究
2015-07-18 02:40韩文玉刘广波李建青吴晋沪
天然气化工—C1化学与化工 2015年6期
韩文玉 ,刘广波 ,王 辉,李建青 ,吴晋沪
(1.中国科学院青岛生物能源与过程研究所,生物燃料重点实验室,山东 青岛 266101;2.中国科学院大学,北京 100049)
六甲基苯(HMB)是一种价格昂贵的精细化学品,可用作材料中间体、医药中间体、有机合成试剂等。早先,六甲基苯主要来源于石油炼制过程中的副产物和五甲基苯、四甲基苯等的甲基化反应。目前,工业上六甲基苯主要由甲醇与苯酚反应制得,反应主要是在固体超强酸等催化剂上进行,此过程中存在工艺复杂,原料毒性大等问题[1]。近期,Sulikowski等[2]以超稳Y分子筛和氧化铝的混合物为催化剂,在 300℃、0.78MPa~2.74MPa的反应条件下催化甲醇反应,制得六甲基苯。Mikkelsen等[3]研究发现HBeta分子筛催化甲醇反应,在较低空速条件下,产物中芳烃主要为六甲基苯。然而,原料毒性、原料利用率低及工艺复杂等缺陷限制了此类方法的应用。
二 甲 醚 分 子 可 提 供 CH3-、CH3O-、CH3OCH2-等基团进行化学反应。随着二甲醚产能的扩增和生产成本的下降,从二甲醚出发开发工业上基于甲醇为原料的高附加值下游化学品受到了越来越多科学家的关注[4-8]。
以二甲醚为原料制备烃类化合物(DTH),机理主要包括:活性烃池物种的生成(多甲基苯类)[9-11];低碳烯烃的生成[12-13];低碳烯烃聚合、环化、氢转移生成烷烃及芳烃等[14-16]。
目前,现有二甲醚制烃类化合物的研究多集中于ZSM-5分子筛催化剂上的反应,受ZSM-5孔道尺寸的限制,产物多为C10以下烃类,芳烃产物主要为甲苯、二甲苯、三甲苯等混合芳烃,这些混合芳烃不易分离,经济价值不高。对混合芳烃化合物进行深度甲基化反应,使其完全甲基化,不仅可以提高产物的经济价值,而且有助于产物分离。通过调节分子筛的孔结构和酸性可以改变其芳构化和甲基化性能,使用改性分子筛催化二甲醚制六甲基苯是一条潜在的提高二甲醚经济价值的路线。
在DTH反应中,催化剂的孔道结构和酸性影响其芳构化和甲基化性能。金属及金属氧化物常用于修饰催化剂的孔结构和调节酸性。Esquivel等[17]研究了过渡金属改性的β分子筛催化甲醇反应性能,发现Mn2+和Zn2+改性有利于提高β分子筛的芳构化性能。王博等[18]研究发现,WO3改性HZSM-5可以催化二甲醚高选择性地转化为甲苯。
目前,关于二甲醚选择性转化制六甲基苯的研究少有报道,本实验采用浸渍法制备了一系列金属改性HBeta分子筛,并考察了不同金属改性HBeta分子筛选择性催化二甲醚制六甲基苯的性能。此外,还考察了反应温度和空速对二甲醚转化制六甲基苯的影响。采用N2物理吸附、NH3-TPD、XRD、吡啶红外技术表征了催化剂的结构和酸性,并分析了不同催化剂的结构和酸性对二甲醚转化制六甲基苯的影响。
1 实验部分
1.1 实验试剂
试剂:HBeta分子筛,硅铝比12.5,南开大学催化剂厂;高纯二甲醚,济宁协力特种气;高纯氮气,南京伟创气体有限公司;偏钨酸铵,Alfa Aesar试剂有限公司;四水合钼酸铵、硝酸钾、六水合硝酸锌、50%硝酸锰溶液,国药集团化学试剂有限公司。
1.2 催化剂制备
采用等体积浸渍法制备MnO2修饰的HBeta催化剂:配制一定浓度的硝酸锰溶液,加入到一定量的HBeta分子筛中,充分搅拌均匀,超声处理2h,再于室温下静置6h,110℃干燥12h,然后于马弗炉中450℃焙烧6h,得MnO2/HBeta分子筛。采用同样方法分别制得 K2O/HBeta、ZnO/HBeta、MoO3/HBeta 和WO3/HBeta,金属氧化物负载质量分数均为10%。所得分子筛粉末经压片、筛分后得40~60目颗粒,待用。
1.3 催化剂性能评价
催化剂的性能评价在固定床反应器内进行,催化剂(40~60目)装填量为9mL,为使原料二甲醚充分预热,催化剂床层上方填装20mL石英砂(40~60目)。反应前催化剂在氮气气氛中400℃原位预处理3h,随后降温至反应温度,通入反应原料二甲醚。反应产物由气、液、固三相组成,气相产物由GC2060气相色谱分析(鲁南分析仪器有限公司,15%邻苯二甲酸二丁酯,6201担体);液、固产物采用安捷伦7820气相色谱分析(安捷伦,色谱柱为DB-1)。
1.4 催化剂的表征
1.4.1 粉末X-射线衍射
XRD在Bruker D8 advance X射线衍射仪上进行,Cu Kα 辐射源,λ=0.152nm,管电压 40kV,管电流 40mA,5~70°扫描, 描速度 2°/min, 扫描步幅0.02°。
1.4.2 氮气物理吸附
样品的比表面积测定在Micromeritics ASAP 2010型物理吸附仪上进行。催化剂首先在450℃下真空脱气处理10h,然后在-196℃进行低温N2物理吸附脱附实验,样品的总比表面积由BET方法求得。
1.4.3 氨程序升温脱附
催化剂的酸性用氨程序升温脱附 (NH3-TPD)法测定,所用仪器为美国Micromeritics Auto ChemⅡ2920吸附仪。催化剂首先在高纯Ar气流(40mL/min)中450℃处理2h,随后降温至100℃吸附φ(NH3)=10%的NH3-Ar混合气 30min至饱和,接着切换为Ar(40mL/min)吹扫1h以除去物理吸附的NH3,最后在 Ar气流下 (40mL/min)以 10℃/min的速率升温至400℃并恒温20min至脱附完全。TCD检测器同步记录信号以测定催化剂的NH3脱附曲线。1.4.4 吡啶吸附红外测试
通过吸附吡啶的红外光谱表征催化剂的Bronsted酸性位和Lewis酸性位,所用仪器为Bruker公司的TENSOR27型红外光谱仪,以吡啶为探针分子,利用吸附吡啶后的特征峰来区分B酸和L酸。准确称取样品15mg压成半透明状直径13mm自支撑圆片,置于红外吸收池中,400℃下高真空净化120min,待红外吸收池温度降至室温后,静态吸附吡啶60min,然后升温至150℃脱附吡啶60min,最后室温下测定红外谱图。
2 结果与讨论
2.1 催化剂筛选
表1为不同金属改性HBeta分子筛催化二甲醚反应性能。由表1可以看出不同金属改性HBeta的催化性能差别较大。未改性的HBeta二甲醚转化率和六甲基苯选择性分别为91.4%和20.3%。与未改性的 HBeta相比,10%MoO3/HBeta和 10%WO3/HBeta的二甲醚转化率未有明显变化,分别为89.3%和90.4%,二者的六甲基苯选择性明显降低,分别为9.9%和8.7%。10%MnO2/HBeta和10%ZnO/HBeta的二甲醚转化率均低于HBeta,分别为76.4%和72.3%,六甲基苯选择性均高于HBeta,分别为25.1%和21.7%。10%K2O二甲醚转化率明显降低,且不具备六甲基苯选择性。由表1可以看出,10%MnO2/HBeta的六甲基苯选择性比10%ZnO/HBeta高。因此,在上述金属氧化物改性HBeta中,10%MnO2/HBeta催化二甲醚制六甲基苯性能最好。
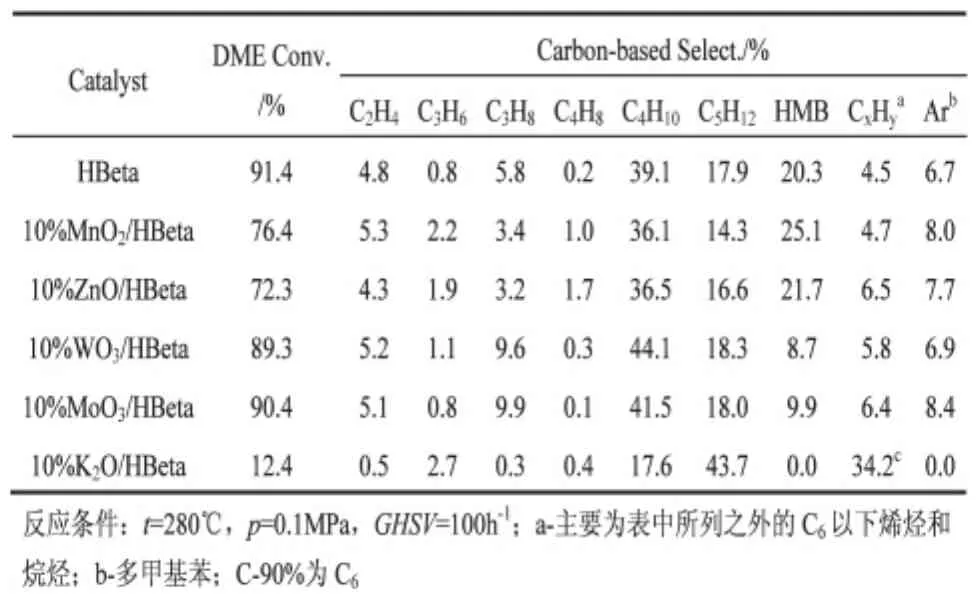
表1 二甲醚在不同催化剂上的反应性能
2.2 反应温度对催化性能的影响
图1为反应温度对10%MnO2/HBeta分子筛催化二甲醚制六甲基苯反应性能的影响。由图1可以看出,随反应温度升高,二甲醚转化率由44.8%逐渐升至96.5%,六甲基苯选择性呈现出先升高后降低的趋势,反应温度为280℃时,六甲基苯选择性最高。Ilias等[19]研究表明,反应温度在280℃以下,MTH最终产物主要为低碳烷烃和环烷烃。由图1可以看出,反应温度小于280℃,随温度降低,丙烷、丁烷和戊烷选择性提高,这与Ilias的研究结论基本一致。Bjorgen等[20-21]研究表明,MTH反应中多甲基苯可以发生脱甲基反应。反应温度较高时,六甲基苯选择性降低,而其它芳烃的总选择性提高,这可能是由于此种条件下多甲基苯脱甲基反应较明显,不利于六甲基苯的生成。由此可以得出结论,反应温度太高、太低均不利于六甲基苯的生成,适宜的反应温度有利于提高六甲基苯的选择性。

图1 反应温度对10%MnO2/HBeta催化二甲醚制六甲基苯反应性能的影响
2.3 反应空速对催化性能的影响
图2为反应空速对10%MnO2/HBeta催化二甲醚制六甲基苯反应性能的影响。由图2可以看出,随反应空速提高,二甲醚转化率不断降低,六甲基苯选择性先提高后降低,反应空速为100h-1时,六甲基苯选择性最高。反应空速低于100h-1时,产物中基本不含丙烯和丁烯;反应空速超过100h-1后,随空速提高,丙烯和丁烯选择性不断提高,这表明空速较高时,低碳烯烃向芳烃的转化不完全,这应该是较高空速下六甲基苯选择性较低的原因。反应空速小于100h-1时,随空速降低,丁烷和戊烷的选择性均大幅提高,而六甲基苯选择性大幅降低。因此,反应空速太低、太高均不利于提高六甲基苯的选择性,适宜的反应空速有利于提高六甲基苯的选择性。
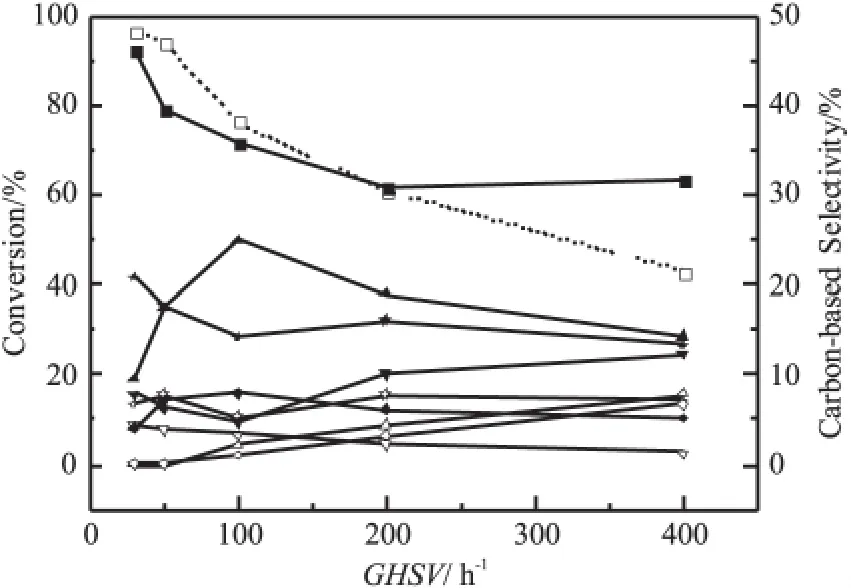
图2 反应空速对10%MnO2/HBeta催化二甲醚制六甲基苯反应性能的影响
2.4 催化剂表征
2.4.1 XRD表征结果
图3为金属改性的HBeta分子筛XRD谱图。由图 3 可知,10%ZnO/HBeta、10%MoO3/HBeta、10%WO3/HBeta、10%K2O/HBeta表现出与HBeta基本一致的谱图结构,这表明上述金属并未明显破坏Beta分子筛的骨架结构,且可能在HBeta分子筛表面均匀分散。10%MnO2/HBeta在37.29°位置出现MnO2的特征衍射峰,10%MnO2/HBeta在43.38°附近不具有衍射峰,而其它催化剂在此位置均具有明显的衍射 峰 ;10%MnO2/HBeta 在 13.37°、14.58°、25.22°、26.98°等处的衍射峰明显弱于其它催化剂,这可能意味着MnO2与HBeta的相互作用强于其它金属氧化物。
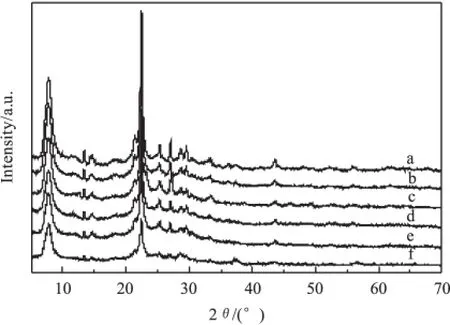
图3 不同催化剂XRD谱图
2.4.2 NH3-TPD表征结果
图4为不同金属改性HBeta分子筛的NH3-TPD表征结果。由图4可以看出,催化剂均呈现出不同强度的强酸、弱酸峰,金属的加入在一定程度上影响了催化剂表面酸性。与HBeta相比,10%MnO2/HBeta具有明显的强酸峰,这说明10%MnO2的加入明显增强了催化剂的强酸中心;10%MnO2/HBeta的六甲基苯选择性较HBeta有所提高,这表明催化剂表面的强酸中心促进六甲基苯的生成。与HBeta相比,10%MnO2/HBeta和10%ZnO/HBeta的弱酸峰向低温区做了相似程度的偏移,这表明10%ZnO和10%MnO2的加入均减弱了催化剂的弱酸中心;10%ZnO/HBeta和10%MnO2/HBeta的二甲醚转化率均较HBeta大幅降低,这表明催化剂表面的弱酸中心有利于二甲醚的转化。与HBeta相比,10%WO3/HBeta和 10%MoO3/HBeta弱酸峰均向低温区有微小偏移,这表明10%WO3和10%MoO3的加入稍微减弱了催化剂的弱酸中心;二者的二甲醚转化率与HBeta相比变化不大,而二者的六甲基苯选择性明显低于HBeta,这表明WO3和MoO3的加入不利于六甲基苯的生成。
2.4.3 红外表征结果
图5为不同催化剂的Py-IR谱图,图中1450cm-1附近为L酸特征峰,1545cm-1附近为B酸特征峰,1490cm-1附近为B酸和L酸共同作用结果。由图5可知,所有催化剂在1450cm-1附近均具有明显的L酸特征峰,这表明上述金属改性HBeta均含有 L酸。与 HBeta相比,10%K2O/HBeta在1545cm-1附近不具有B酸特征峰,这表明10%K2O/HBeta不含B酸;HBeta具有六甲基苯选择性,而10%K2O/HBeta不具有六甲基苯选择性,这表明B酸是生成六甲基苯所必须的。与HBeta相比,10%WO3/HBeta和10%MoO3/HBeta的B酸峰均强于HBeta,这表明二者B酸多于 HBeta;10%WO3/HBeta和10%MoO3/HBeta的六甲基苯选择性均小于HBeta,这表明B酸太多不利于六甲基苯的生成。与 HBeta相比,10%MnO2/HBeta和 10%ZnO/HBeta的B酸峰弱于HBeta,这表明10%MnO2/HBeta和10%ZnO/HBeta的B酸少于HBeta;10%MnO2/HBeta和10%ZnO/HBeta的六甲基苯选择性高于HBeta,这表明适度降低B酸量有利于六甲基苯的生成。B酸含量过低或过高均不利于六甲基苯的生成。

图4 不同催化剂的NH3-TPD谱图
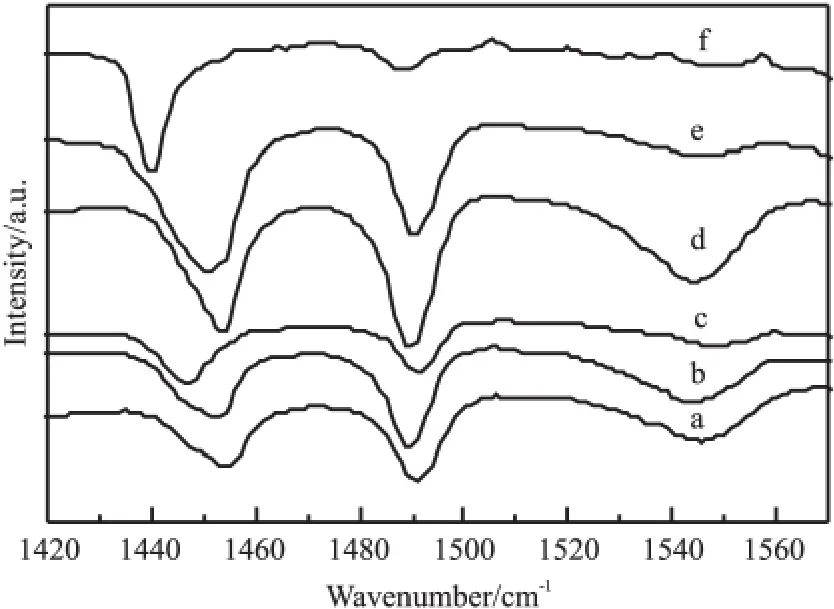
图5 不同催化剂的Py-IR谱图
2.5 六甲基苯生成机理
研究表明MTH反应中,二甲醚在B/L酸作用下生成低碳烯烃[16],低碳烯烃在B酸作用下环化脱氢生成环烷烃和多烯烃[12,22],环烷烃和多烯烃在B/L酸作用下生成芳烃和链烷烃,芳烃与甲醇/二甲醚在B/L酸作用下发生甲基化反应生成多甲基取代苯[16,23]。本实验中含有L酸而不含B酸的10%K2O/HBeta不具有芳烃选择性,可能是由于缺少B酸的作用低碳烯烃无法环化所致。另外,B酸量高于HBeta的10%MoO3/HBeta六甲基苯选择性低于HBeta,C3~C5烷烃的选择性高于 HBeta;而 B 酸量低于HBeta的10%MnO2/HBeta,其六甲基苯选择性高于HBeta,C3~C5烷烃的选择性低于HBeta,这可能由于B酸催化环烷烃和多烯烃生成芳烃的同时通过氢转移生成了烷烃,而L酸催化环烷烃和多烯烃生成芳烃时直接生成了H2,使得B酸含量较高时,芳烃选择性降低,进而降低了六甲基苯的选择性。根据已有文献和本实验结果,对二甲醚转化制六甲基苯机理进行推测,可能的反应机理示意图如图6所示。

图6 二甲醚制六甲基苯反应机理
3 结论
考察了 K、Mn、Mo、W、Zn 改性 HBeta对催化二甲醚制六甲基苯性能的影响,结果表明,10%MnO2/HBeta具有较高的六甲基苯选择性。另外,还在10%MnO2/HBeta上考察了反应温度和空速对六甲基苯选择性的影响,结果表明,六甲基苯选择性随反应温度升高呈现先升高后降低的趋势,最佳反应温度为280℃;六甲基苯选择性随反应空速的升高先升高后降低,最佳反应空速为100h-1。
不同金属改性催化剂BET、NH3-TPD、Py-IR表征结果表明,比表面积降低不利于二甲醚的转化,适宜的强酸量有利于六甲基苯的生成;单独的L酸可催化二甲醚反应,但不具有六甲基苯选择性,适宜的B酸量有利于六甲基苯的生成。
[1]高根之.一种用颗粒型固体超强酸催化合成六甲基苯的方法[P].CN:201410198124.5,2014.
[2]Sulikowski B,Popielarz A.Conversion of methanol on ultrastable faujasitic catalysts-selective formation of hexamethylbenzene[J].Appl Catal,1988,42:195-203.
[3]Mikkelsen Φ,Kolboe S.The conversion of methanol to hydrocarbons over zeolite HBeta [J].Microporous Mesoporous Mater,1999,29:173-184.
[4]Zhang Q D,Tan Y S,Liu G B,et al.Promotional effects of Sm2O3on Mn-H4SiW12O40/SiO2catalyst for dimethyl ether direct-oxidation to dimethoxymethane[J].J Ind Eng Chem,2014,20:1869-1874.
[5]刘广波,张清德,寇永利,等.二甲醚氧化制甲酸甲酯MoSnO催化剂再生性能研究[J].天然气化工(C1化学与化工),2013,38(4):1-5.
[6]Xue H F,Huang S M,Ditzel E,et al.Dimethyl ether carbonylation to methyl acetate over nanosized mordenites[J].Ind Eng Chem Res,2013,52:11510-11515.
[7]Luft G,Ritter G,Schrod M.Homogeneously catalyzed carbonylation of dimethyl ether to acetic-anhydride[J].Chem IngTech,1982,54:758-760.
[8]Tkachenko O P,Kucherov A V,Glukhov L M,et al.Spectral studies of catalysts of oxidative dehydrogenation of dimethyl ether to dimethoxyethane[J].Russ J Phy Chem A,2013,87:1249-1251.
[9]Dahl I M,Kolboe S.On the reaction mechanism for hydrocarbon formation from methanol over SAPO-34(I):Isotopic labeling studies of the co-reaction of ethene and methanol[J].J Catal,1994,149:458-464.
[10]Mikkelsen Φ,RΦnning P O,Kolboe S.Use of isotopic labeling for mechanistic studies of themethanol-tohydrocarbons reaction:Methylation oftoluene with methanol over H-ZSM-5,H-mordenite and H-beta[J].Microporous Mesoporous Mater,2000,40:95-113.
[11]Song W,Marcus D M,Fu H,et al.An oft-studied reaction that may never have been:Direct catalytic conversion of methanol or dimethyl ether to hydrocarbons on the solid acids HZSM-5 or HSAPO-34[J].J Am Chem Soc,2002,124:3844-3845.
[12]Hemelsoet K,Van Der Mynsbrugge J,De Wispelaere K,et al.Unraveling the reaction mechanisms governing methanol-to-olefins catalysis by theory and experiment[J].Chem Phys Chem,2013,14:1526-1545.
[13]Arstad B,Nicholas J B,Haw J F.Theoretical study of the methylbenzene side-chain hydrocarbon pool mechanism in methanol to olefin catalysis[J].J Am Chem Soc,2004,126:2991-3001.
[14]Olsbye U,Svelle S,Bj?rgen M,et al.Conversion of methanol to hydrocarbons:how zeolite cavity and pore size controls product selectivity[J].Angew Chem Int Edit,2012,51:5810-5831.
[15]Olsbye U,BjΦrgen M,Svelle S,et al.Mechanistic insight into the methanol-to-hydrocarbons reaction[J].Catal Today,2005,106:108-111.
[16]Haw J F,Song W G,Marcus D M,et al.The mechanism of methanol to hydrocarbon catalysis[J].Accoun Chem Res,2003,36:317-326.
[17]Esquivel D,Cruz-Cabeza A J,Jiménez-Sanchidrián C,et al.Transition metal exchanged β zeolites:Characterization of the metal state and catalytic application in the methanolconversion to hydrocarbons[J].Microporous Mesoporous Mater,2013,179:30-39.
[18]王博,王辉,刘广波,等.二甲醚催化转化制甲苯的反应研究[J].燃料化学学报,2014,42(8):994-1000.
[19]Ilias S,Bhan A.Mechanism of the catalytic conversion of methanol to hydrocarbons[J].Acs Catal,2013,3:18-31.
[20]BjΦrgen M,Olsbye U,Kolboe S,et al.Coke precursor formation and zeolite deactivation:mechanistic insights from hexamethylbenzene conversion[J].J Catal,2003,215:30-44.
[21]Ilias S,Bhan A.The mechanism of aromatic dealkylation in methanol-to-hydrocarbonsconversion on H-ZSM-5:What are the aromatic precursors to light olefins[J].J Catal,2014,311:6-16.
[22]张宝珠.甲醇转化制芳烃(MTA)反应的研究[D].大连:大连理工大学,2013.
[23]Svelle S,Visur M,Olsbye U,et al.Mechanistic aspects of the zeolite catalyzed methylation of alkenes and aromatics with methanol:A review[J].Top Catal,2011,54:897-906.
猜你喜欢
化工管理(2022年13期)2022-12-02
水泵技术(2022年3期)2022-08-26
汽车实用技术(2022年9期)2022-05-20
航空维修与工程(2022年11期)2022-02-06
煤气与热力(2021年11期)2021-12-21
石油沥青(2021年4期)2021-10-14
科学家(2021年24期)2021-04-25
缔客世界(2020年10期)2020-04-10
中国新技术新产品(2017年3期)2017-03-07
浙江大学学报(工学版)(2016年2期)2016-06-05
