
儿童多发性大动脉炎1例报道并文献复习
2015-05-24 16:22:18李晶芦惠
浙江中西医结合杂志 2015年8期
李晶芦惠
儿童多发性大动脉炎1例报道并文献复习
李晶芦惠
儿童;多发性大动脉炎
多发性大动脉炎(takayasu’s arteritis,TA)是一种少见的慢性大血管炎症,主要累及主动脉及其分支和肺动脉,其病因不明,感染、遗传、自身免疫均在疾病发生中起重要作用。临床表现不典型,极易误诊、漏诊,甚至造成心力衰竭、肾功能衰竭、猝死等严重后果。本文报道1例临床表现极不典型而被成功诊治的多发性大动脉炎患者,同时结合文献综述对该病的诊断、治疗进行分析,以指导临床。
1 临床资料
1.1 病史与查体 患儿女性,13岁,因“反复头痛2年,发现血压高1天”入院。2年前患儿出现反复头痛,休息后自行缓解,曾诊断“血管性头痛”,未予特殊治疗,无呕吐、发热、惊厥,1天前因体检发现血压高(170/100mmHg)入院。否认高血压、心脏病、糖尿病史,否认高血压家族史。查体:体温37.4℃,脉搏106次/分,呼吸19次/分,BP 226/129mmHg,四肢血压:右上肢216/136mmHg,左上肢227/145mmHg,右下肢163/114mmHg,左下肢176/126mmHg。神清,精神可,皮肤无红斑及硬结,颈软,心界无扩大,心律齐,心音中等,未闻及病理性杂音,腹软,背部及腹壁可闻及2/6~3/6级吹风样杂音,双侧肱动脉及足背动脉博动有力,克氏征、布氏征阴性,双侧巴氏征阴性。眼底检查:A:V=1:2,眼底未见渗出。
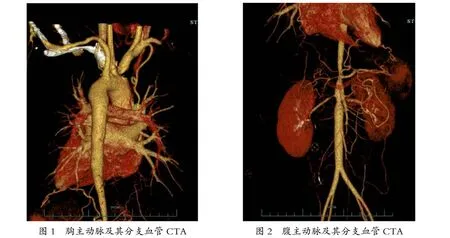
1.2 实验室检查 血沉46mm/h,抗O、CRP正常,结核杆菌抗体及T-SPOT检查均阴性,自身抗体阴性。心脏超声检查:左房内隔膜回声(未致明显血流动力学改变),心率偏快,余心脏形态、结构及瓣膜活动未见明显异常。大动脉超声检查:腹主动脉内膜弥漫性增厚、管腔狭窄,双侧肾动脉起始处内膜回声增强。头颅CT平扫未见明显异常。胸腹血管CTA增强扫描:腹主动脉节段性管壁增厚,管腔狭窄,腹腔干开口水平重度狭窄,腹腔干、左肾动脉开口受累,重度狭窄,肠系膜上动脉及右侧肾动脉开口处轻中度狭窄(封二,图1~2)。
2 诊断和治疗
根据2008年安卡拉会议提出的最新诊断标准[1],确诊为多发性大动脉炎,予强的松1mg/(kg·d)口服,硝苯地平、利尿剂口服降压,好转出院。后至其他医院治疗,加用甲氨蝶呤12.5mg口服,1周1次,缓慢减少强的松剂量,目前强的松减量至0.3mg/(kg·d),隔日顿服。随访2年患儿生命体征稳定,血压控制在130~140/70~90mmHg,定期复查CRP、血沉均在正常范围,复查血管CTA无新发病灶。
3 讨论
该病例具有以下特点:①临床表现不典型,仅表现为反复头痛,而无低热、跛行等表现;②体征较为典型,体检可发现高血压、四肢血压差、血管杂音;③辅助检查较为典型,血沉增快,大动脉超声及大动脉CTA增强扫描均支持该诊断。
3.1 临床表现及诊断 该病临床表现多不典型,主要有发热、乏力、体质量减低、上下肢脉压差增大、无脉等。TA可引发多种严重并发症,如肺动脉血栓形成、脑梗塞、视力下降甚至失明。据报道的30%~50%的患者缺乏全身症状[2-3]。
TA诊断需依靠临床表现、体格检查、实验室检查及影像学检查综合判断。目前儿童最新的诊断标准是2008年在安卡拉会议上提出:主动脉及其分支血管造影异常为必备条件,同时符合以下5项标准中的任何1项可诊断:①脉搏减弱或消失;②四肢血压差异常;③血管杂音;④高血压;⑤急性期炎症反应,包括血沉>20mm/h或C反应蛋白升高[1]。本例患者除无脉搏变化外,余指标均符合,故TA诊断明确。
对于血管病变发生前的初期患者的早期诊断较为困难。建议对于40岁以下存在下列症状的患者均需考虑有TA可能[4]:①不明原因的急性期反应物升高(CRP或血沉,或两者均有);②高血压;③双上肢血压差>10mmHg;④脉搏减弱或消失;⑤跛行;⑥血管杂音。需动态监测影像学检查。
TA的分类方法较多,国际上通用的方法是1996年由Numano组制定的六分类法[5],该分类方法需基于血管造影分类。本例患者血管病变累及腹主动脉和肾动脉,依据六分类法,本例属Ⅳ型。
3.2 影像学检查及活动性标志物 影像学检查在TA的诊断中起着重要作用。血管造影为TA诊断金标准,但不是首选检查,而是作为MRA和CTA检查的补充;MRA能更好的显示血管壁的变化,早期提示炎症活动,但对血管远端分支显示欠准确,故常用于早期诊断及长期随访;CTA可以发现血管的早期病变,而且在评估TA是否活动可能比血清学检查更敏感[6],常用于疾病中期及进展期的诊断;高分辨率超声适用于长期复查随访;氟—氟脱氧葡萄糖正电子发射CT(F18-FDG-PET)可以较敏感发现病变的位置并估计炎症的活动强度。Lee等[7]也发现F18-FDG-PET检测值与CRP、ESR成正相关,可敏感判定TA活动期。但F18-FDG-PET辐射大、费用高,目前尚未广泛应用。
TA的炎症活动可引起受累血管腔进一步狭窄。目前国际上推荐综合了临床表现、实验室检查及影像学表现的评分法判断活动期。通常应用的评分法为美国国立研究院(national institutes of health,NIH)评分法[7]:①全身症状如发热、肌肉骨骼酸痛(除外其他疾病);②红细胞沉降率升高;③血管炎的典型表现,如跛行、脉搏减弱、血管杂音、血管疼痛(颈动脉痛)、四肢血压差异;④血管造影检查。上述4项中2项以上为新发或加重则提示血管炎活动期。最近Misra等[8]提出了新的评分方法(ITAS 2010),目前该评分标准尚未大范围应用,仍在一些临床中心观察研究。
3.3 治 疗 TA包括药物治疗和手术治疗。糖皮质激素是TA治疗的一线用药。约60%~80%的患者单用激素即可得到缓解[7]。约50%的患者在激素减量过程中出现复发[9]。免疫抑制剂常用于激素耐药或激素减量中复发者,目前甲氨蝶呤因其副作用小且效果较好,目前成为临床最常用的免疫抑制剂。
EULAR建议[10]强的松初始剂量1mg/(kg·d),最大剂量60mg/d。病危者可用大剂量甲泼尼龙冲击治疗,常用剂量为1次15~30mg/kg(日最大剂量应<1g),连用3天为1个疗程[11]。最初治疗3个月时皮质激素剂量不应少于10~15mg/d,减量期间不主张改为隔天应用,因为可致疾病复发[10]。待炎症指标控制后(即血沉和CRP趋于正常,常需4周或更长时间)开始逐渐减量,至5~10mg隔天1次维持。甲氨蝶呤可口服0.3mg/(kg·w)或5~25mg/w[11],常于用药第2天补充叶酸5mg以减轻不良反应,注意监测血、尿常规及肝肾功能。
生物制剂的靶向治疗目前在免疫系统疾病的应用越来越多,且疗效得到肯定,是未来的治疗方向。肿瘤坏死因子-抑制剂代表药物即英夫利昔单抗,在儿童TA的治疗中较环磷酰胺有更高的安全性和有效性[12]。托珠单抗为IgG亚类的重组人源化IL-6受体单克隆抗体,在难治性TA治疗中可使病情得到一定的控制,激素剂量得以减低。
外科治疗的目的是使缺血部位重建血流供应。需在病情稳定后进行,手术治疗仅能恢复血液供应,而不能阻断疾病进展。TA的预后主要取决于炎症持续状态及并发症的进展。儿童TA的病死率高达35%,早期诊断、早期治疗,能改善预后。
综上所述,TA在儿童中的病死率较高,因缺乏特征性表现故早期诊断较困难。对于高血压儿童应详细体格检查,尽量做到早期诊断,早期治疗,以提高存活率和生存质量。
[1]Ozen S,Pistorio A,Iusan SM,et al.EULAR/PRINTO/PRES criteria for Henoeh-Schnlein purpura,childhood polyarteritis nodosa,childhood Wegener granulomatosis and childhood Takayasu arteritis:Ankara 2008.Part II:Final classification criteria[J].Ann Rheum Dis,2010,69(5):798-806.
[2]Maksimowicz-McKinnon K,Clark T,Hoffman G,et al.Limitations of therapy and a guarded prognosis in an American cohort of Takayasu arteritis patients[J].Arthritis Rheum,2007,56(3):1000-1009.
[3]Park M,Lee S,Park Y,et al.Clinical characteristics and outcomes of Takayasu’s arteritis:analysis of 108 patients using standardized criteria for diagnosis,activity assessment,and angiographic classification[J].Scand JRheumatol,2005,34(4):284-292.
[4]Mason JC.Takayasu arteritis-advances in diagnosis and management[J].Rheumatology,2010,6(7):406-415.
[5]Hata A,Noda M,Moriwaki R,et al.Angiographic findings of Takayasu arteritis:new classification[J].Int JCardiol,1996,54(1):155-163.
[6]Khandelwal N,Kalra N,Garg MK,et al.Multidatector CT angiography in Takayasu arteritis[J].Eur J Radid,2011,77(2):369-374.
[7]Kerr GS,Hallahan CW,Giordano J,et al.Takayasu arteritis[J].Ann InternMed,1994,120(11):919-929.
[8]Misra R,Danda D,Rajappa SM,et al.Development and initial validation of the Indian Takayasu Clinical Activity Score(ITAS2010)[J].Rheumatology,2013,52(10):1795-1801.
[9]Seibel S,Gaa J,Küchle C,et al.Severe renovascular hy pertension in apatientwith Takayasu arteritis[J].Am JKidney Dis,2010,56(3):595-598.
[10]Mukhtyar C,Guillevin L,Cid MC,et al.EULAR recom mendations for the management of large vessel vasculitis[J]. Ann Rheum Dis,2009,68(3):318-323.
[11]中华医学会风湿病学分会.大动脉炎诊断及治疗指南[S].中华风湿病学杂志,2011,15(2):119-120.
[12]Stern S,Clemente G,Reiff A,et al.Treatment of Pediatric Takayasu arteritis with infliximab and cyclophosphamide:experience from an American-Brazilian cohort study[J]. Clin Rheumatol,2014,20(4):183-188.
(收稿:2014-12-05 修回:2015-02-04)
杭州市第一人民医院儿科(杭州 310006)
芦惠,Tel:13575789528;E-mail:luhui6699@sina.com
猜你喜欢
青春期健康·青少版(2023年4期)2023-06-25 15:26:57
青春期健康(2023年8期)2023-06-19 06:51:58
公民与法治(2022年6期)2022-07-26 06:16:02
中国临床医学影像杂志(2021年10期)2021-11-22 07:46:50
中华养生保健(2020年7期)2020-11-16 01:14:28
心肺血管病杂志(2019年1期)2019-04-22 01:12:08
高中生·天天向上(2017年4期)2017-06-09 02:37:17
中国心血管杂志(2016年6期)2017-01-09 07:20:34
中国医疗美容(2015年1期)2015-07-12 10:06:04
现代检验医学杂志(2014年4期)2014-02-02 02:44:51
