
基于HBV序列的系统进化分析和特征提取研究
2015-04-18 03:00谢坐祥张俊鹏
大理大学学报 2015年12期
谢坐祥,陈 霞,张俊鹏
(大理大学工程学院,云南大理 671003)
乙型肝炎病毒(Hepatitis B Virus,HBV)是一类引起人类慢、急性肝炎的环状DNA病毒〔1〕。目前全球约有2.4亿人感染乙型肝炎,每年约有78万人死于慢性或急性乙型肝炎。根据世界卫生组织(WHO)〔1〕报告,我国有5%~10%的成年人是HBV慢性感染者。
HBV基因为部分闭合双链环状DNA,全长约3.2 kb。它主要分为P、X、C和S 4个基因区,C区分为C基因和前C基因片段,S区分为前S1、前S2和S基因片段〔2〕。目前研究表明:HBV基因型可以分为A、B、C、D、E、F、G和H 8种类型,不同的基因型呈现不同地理区域分布,我国主要以B和C两种基因型为主〔3〕。
本文将基于机器学习算法和数据挖掘技术实现HBV多序列比对、系统进化分析和特征提取3个层次的分析。这将有利于进一步了解HBV病毒在序列层次下的进化关系、突变过程、基因特点和基因型种类,进而为HBV患者提供更科学有效的辅助治疗。
1 材料与方法
1.1 数据源 本文选用的HBV序列数据源来源于云南省第一人民医院〔3〕,选取的HBV序列片段为X和前C基因片段。10例HBV感染者样本使用聚合酶链式反应(Polymerase Chain Reaction,PCR)扩增技术克隆至364个样本,每例样本的克隆数如表1所示。

表1 HBV感染者样本及其克隆数
1.2 分析方法 HBV序列分析流程图如图1所示,整个分析过程由3个步骤组成。步骤1获取HBV数据源,数据源包括364个HBV样本序列和38个HBV参考序列,然后进行多序列比对。为了保证HBV多序列比对的质量,本文采用手动比对方法对HBV序列进行多序列比对。步骤2就比对后的HBV序列进行系统进化分析,通过构造系统进化树建立HBV样本序列与参考序列之间的进化分析。本文使用邻接法〔4〕、最大似然法〔5〕、最小进化法〔6〕、平均距离法〔7〕和最大简约法〔8〕5种常用方法分别对10例HBV克隆序列构建系统进化树;为了研究碱基位点与HBeAg阳性与阴性的关系,步骤3对364个HBV样本序列进行特征碱基位点提取,提取方法采用CFS(Correlation Feature Selection)〔9〕、卡方检验(Chi-square Test)〔10〕和信息熵(Information Entropy)〔11〕3种方法进行特征提取。为了评价特征提取前后的分类精度,分别使用决策树C4.5算法〔12〕、朴素贝叶斯(Nave Bayes)〔13〕、支持向量机(SVM)〔14〕和随机森林(Random Forest)〔15〕4种分类器对HBV序列样本进行分类精度比较。
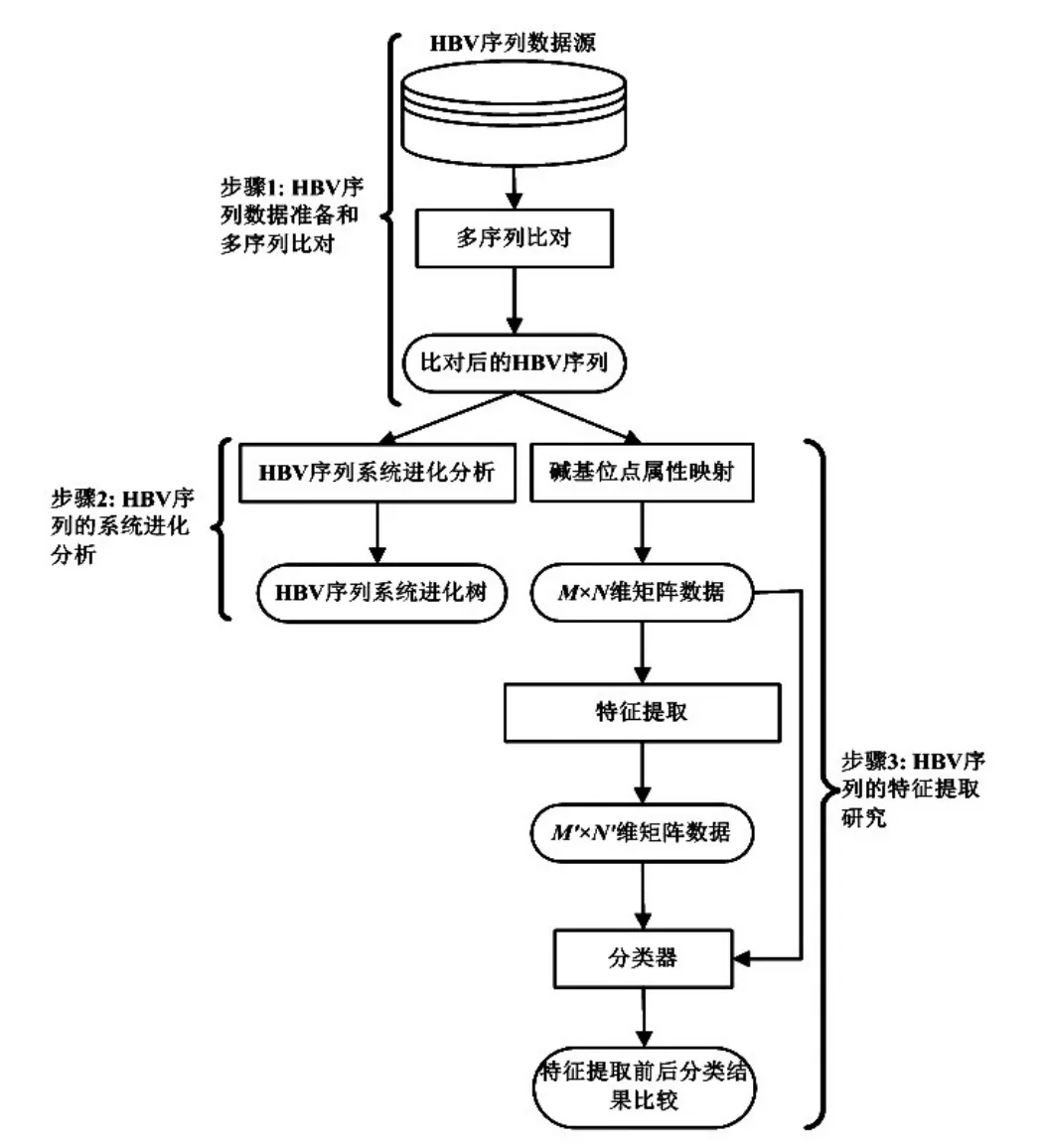
图1 HBV序列分析流程图
2 HBV序列系统进化分析
进化分析是从分子特性出发了解生物系统的内在规律。进化论表明物种之间存在一定的亲缘关系,一般用系统进化树距离的大小表示序列进化中亲缘关系的远近。
本文使用MEGA v6软件中的邻接法、最大似然法、最小进化法、平均距离法和最大简约法5种方法对10例HBV克隆序列构建系统进化树,参数为MEGA软件默认设置参数。通过分析系统进化树,5种方法所预测的HBV基因型完全一致,这也验证了不同方法预测HBV基因型结果一致性。
除了编号8、13和24的真实基因型未知外,其余编号的预测基因型与真实基因型结果完全一致,见表1。这也进一步说明采用系统进化树预测未知HBV序列基因型的方法是有效的。
3 HBV序列特征提取
特征提取也叫特征选择,它是数据预处理的关键步骤。其主要思想是从已知的特征属性集中根据某一特定准则提取出区分性较好的单个特征子集或一个最优特征属性子集〔16〕。经特征提取后可以剔除大量的冗余信息和不相关特征信息,这不仅降低特征属性空间维数,还节约分析时间和提高对目标函数的预测性能力。
目前研究表明,每条HBV序列的碱基位点中,往往很多碱基位点是保守的,与HBV基因型分类无关,因此对HBV序列的碱基位点进行特征提取可以提高HBV基因型正确率和预测水平,同时在序列层次下挖掘与乙型肝炎患者相关的单核苷酸多态性位点(Single-Nucleotide Polymorphism,SNP)〔17〕。
3.1 特征碱基位点提取 HBV序列经过多序列比对后,总共有624个碱基位点。由于CFS方法提取的是一个最优属性集,其大小为11。然而,卡方和信息熵方法按照权重重要性排列每个碱基位点,其大小为624。为了公平地比较他们之间的分类精度,卡方和信息熵方法都统一选择前11个最具有代表性的特征属性集。
另一方面,为了研究属性集大小与分类精度之间的关系,将卡方和信息熵方法提取的特征属性集大小扩大至20、30、40和50。
3.2 分类结果分析 本文选取决策树C4.5、Naïve Bayes、SVM和Random Forest 4种经典分类器对CFS、卡方和信息熵3种特征提取前后的HBV序列进行分类分析。软件平台为WEKA v3.7,属性集大小设置为11,20、30、40和50。
如表2所示,Original代表原始HBV序列数据,CFS-11代表CFS特征提取方法后的HBV序列数据,Chi-11、Chi-20、Chi-30、Chi-40和Chi-50分别代表卡方特征提取法的前11、20、30、40和50特征属性集大小的HBV序列数据,InfoGain-11、InfoGain-20、InfoGain-30、InfoGain-40和InfoGain-50分别代表信息熵特征提取法的前11、20、30、40和50特征属性集大小的HBV序列数据。当特征属性集大小为11的时候,4种分类器的分类精度ACC(Accuracy)有所降低,但是所选择的11个特征属性集也能够很好的表征出原始HBV序列数据的624个特征属性。随着特征属性集大小的增大,4种分类器的分类精度ACC都有增大的趋势。特别地,当特征属性集大小选择合适时,Naïve Bayes和Random Forest的分类精度ACC可以达到最大值1。这些结果表明:特征提取对HBV序列数据降维的同时,也能够保证甚至提高分类精度ACC。
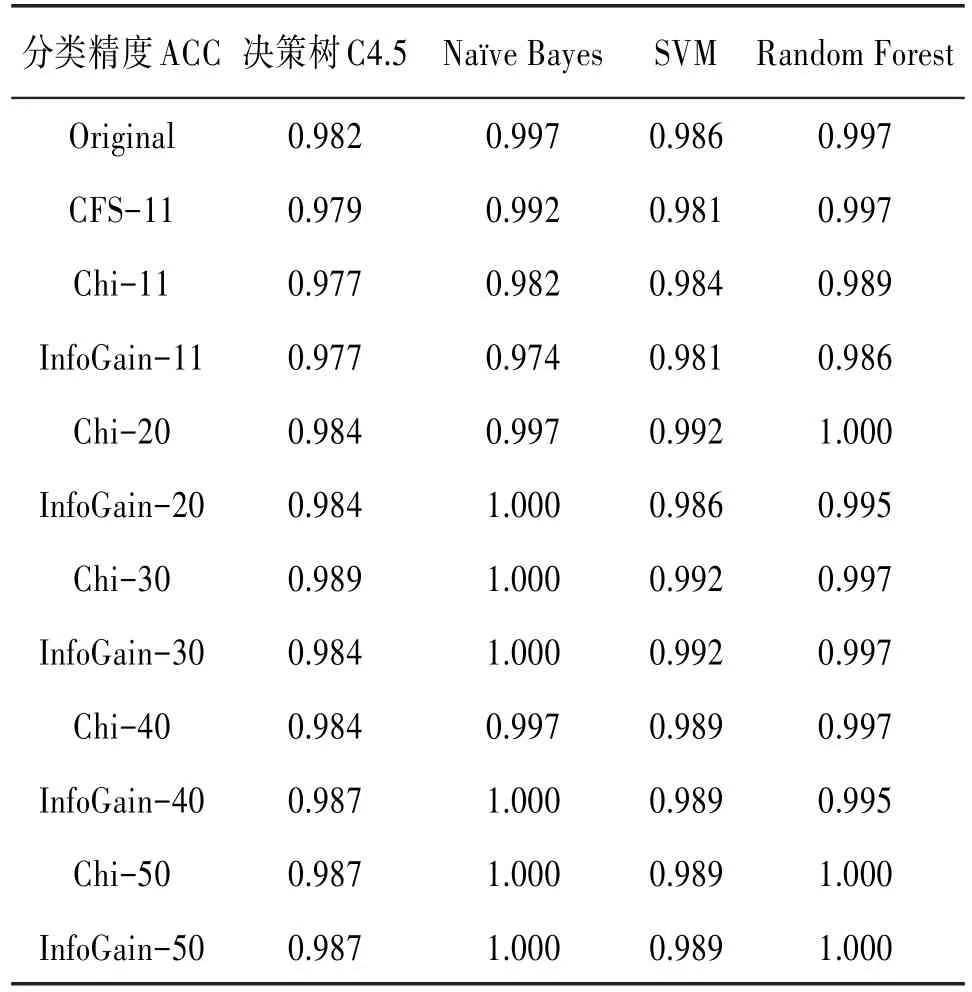
表2 比较不同特征提取方法的分类精度ACC
4 结语
本文从多序列比对、系统进化分析和特征提取3个层次对10例HBV感染者的HBV序列进行分析。首先采用手动比对方法对HBV序列进行多序列比对。然后利用邻接法、最大似然法、最小进化法、平均距离法和最大简约法构造10例HBV克隆序列的系统进化树。5种系统进化树构造法的预测结果完全一致,预测的10例(编号8、10、13、17、24、26、32、213、264和320)HBV感染者基因型分别为:C、C、C、Ba、C、C、C、C、C和C。除去未知基因型,准确率为100%。为了降低HBV序列数据的维数,采用CFS、卡方检验和信息熵3种方法进行特征提取。4种分类器(决策树C4.5、Naïve Bayes、SVM和Random Forest)的分类结果表明:特征提取能够降低HBV序列数据的维数,同时保证甚至提高分类精度。
〔1〕World Health Organization.乙型肝炎实况报道第204号〔EB/OL〕.〔2015-07-19〕.http://www.who.int/mediacentre/factsheets/fs204/zh/.
〔2〕BRECHOT C,POURCEL C,LOUISE A,et al.Presence of integrated hepatitis B virus DNA sequences in cellular DNA of human hepatocellular carcinoma〔J〕.Nature,1980,286(5772):533-535.
〔3〕SHEN T,GAO J,ZOU Y L,et al.Novel hepatitis B virus subgenotype in the southern Yunnan Province of China〔J〕.Intervirology,2009,52(6):340-346.
〔4〕SAITOU N,NEI M.The neighbor-joining method:a new method for reconstructing phylogenetic trees〔J〕.Molecular Biology and Evolution,1987,4(4):406-425.
〔5〕YANG Z.PAML:a program package for phylogenetic analysis by maximum likelihood〔J〕.Computer Applications in the Biosciences:CABIOS,1997,13(5):555-556.
〔6〕RZHETSKY A,NEI M.A simple method for estimating and testing minimum-evolution trees〔J〕.Mol Biol Evol,1992,9(5):945-967.
〔7〕TAKEZAKI N,NEI M.Genetic distances and reconstruction of phylogenetic trees from microsatellite DNA〔J〕.Genetics,1996,144(1):389-399.
〔8〕SAITOU N,IMANISHI T.Relative efficiencies of the Fitch-Margoliash,maximum-parsimony,maximum-likelihood,minimum-evolution,and neighbor-joining methods of phylogenetic tree construction in obtaining the correct tree〔J〕.Mol Biol Evol,1989,6(5):514-525.
〔9〕HALL M A.Correlation-based feature selection for machine learning〔D〕.Hamilton:The University of Waikato,1999.
〔10〕YATES F.Contingency tables involving small numbers and the χ2test〔J〕.Supplement to the Journal of the Royal Statistical Society,1934,1(2):217-235.
〔11〕KULLBACK S,LEIBLER R A.On information and sufficiency〔J〕.The Annals of Mathematical Statistics,1951,22(1):79-86.
〔12〕QUINLAN J R.C4.5:programs for machine learning〔M〕.Amsterdam:Elsevier,2014.
〔13〕RISHI.AnempiricalstudyofthenaiveBayesclassifier〔C〕//IJCAI 2001 workshop on empirical methods in artificial intelligence.2001,3(22):41-46.
〔14〕CORTES C,VAPNIK V.Support-vector networks〔J〕.Machine Learning,1995,20(3):273-297.
〔15〕 BREIMAN L.Random forests〔J〕.Machine Learning,2001,45(1):5-32.
〔16〕GUYON I,ELISSEEFF A.An introduction to variable and feature selection〔J〕.The Journal of Machine Learning Research,2003,3:1157-1182.
〔17〕AHMADIAN A,GHARIZADEH B,GUSTAFSSON A C,et al.Single-nucleotide polymorphism analysis by pyrosequencing〔J〕.Analytical Biochemistry,2000,280(1):103-110.
猜你喜欢
天津市教科院学报(2021年5期)2021-11-10
生物学通报(2021年9期)2021-07-01
教学考试(高考生物)(2020年6期)2020-11-23
食品与生物技术学报(2020年8期)2020-01-06
电子制作(2019年15期)2019-08-27
学苑创造·B版(2019年5期)2019-06-14
科学24小时(2019年5期)2019-06-11
电子制作(2018年19期)2018-11-14
自动化学报(2017年11期)2017-04-04
中国医学创新(2017年7期)2017-03-31
