
锂电池正极材料LiFePO4的电子结构和晶形模拟研究
2015-03-23 04:04黄辉胜胡武洪张国庆张福兰丁世敏
原子与分子物理学报 2015年5期
黄辉胜, 胡武洪, 张国庆, 张福兰, 丁世敏
(1.重庆市无机特种功能材料重点实验室, 重庆 408100; 2.长江师范学院化学化工学院, 重庆 408100)
锂电池正极材料LiFePO4的电子结构和晶形模拟研究
黄辉胜, 胡武洪, 张国庆, 张福兰, 丁世敏
(1.重庆市无机特种功能材料重点实验室, 重庆 408100; 2.长江师范学院化学化工学院, 重庆 408100)
分别运用Materials Studio软件的CASTEP和Morphology模块对磷酸铁锂(LiFePO4)的结构和晶形进行计算模拟研究,获得其优化构型、电子结构、晶体形貌、晶面面积和晶面相对生长速率等特征参数.研究结果表明,PBE方法的计算精度明显高于CA-PZ方法,化合物中的Fe—O配位键本质上是共价键.LiFePO4的理论模拟晶形为柱状晶体,与其块状的实测晶体形貌存在较大差异,这归因于理论模拟晶形是LiFePO4在真空中的生长晶习,而非真实环境下的晶习.在LiFePO4的实际晶形中,(0 2 0)晶面拥有较大的面积,这与该晶面的原子排列方式和原子占有率密切相关.此外,表面活性剂和LiFePO4各特征晶面之间可形成大量C—H…O氢键,将晶粒有效地包覆起来,降低其生长速率,从而抑制LiFePO4晶粒长大.
LiFePO4; 电子结构; 晶形; 溶剂效应; 表面活性剂
1 引 言
橄榄石型LiFePO4自1997年被Padhi等[1]报道以来,因其比容量高、热稳定性和循环性能良好、成本低廉、环境友好等优点而被视为最具发展前景的锂离子二次电池正极材料.但是,LiFePO4也具有两个比较明显的缺点[2],一是室温条件下的电子电导率和锂离子扩散率低,制约了高倍率下的放电性能;二是制备过程中Fe2+很容易被氧化成Fe3+,从而引入非电化学活性杂质.针对这些缺陷,科研人员做了大量的改进工作.其中,在制备过程中加入表面活性剂,得到颗粒细小均匀且形貌规则的LiFePO4是一种比较常见的改性方法[2,3].
众所周知,即使是具有相同分子结构的物质,在不同的外界条件下也可能拥有不同的晶体形貌.因此决定物质晶形的因素主要有两个:一是晶体自身的分子结构,二是晶体所处的外部环境[4].对于外部因素来说,主要包括温度、溶剂和表面活性剂等.近年来,关于磷酸铁锂的结构和晶形已有大量文献报道[5-10].但有关溶剂和表面活性剂究竟如何影响LiFePO4的晶粒尺寸和形貌的研究尚未见报道.因此,本文主要针对LiFePO4的微观结构和理论晶形、以及影响其晶形的内在因素和外部因素进行较深入的理论研究,以揭示表面活性剂控制晶粒形貌和尺寸的微观作用机理.
2 计算方法
LiFePO4的晶体结构取自X射线衍射数据,其晶胞结构如图1所示[5],该晶体属于正交晶系Pnma空间群,每个晶胞含有4个最小不对称单元,其晶胞参数为a=10.227 Å、b=6.005 Å、c=4.692 Å、α=β=γ=90°.首先以LiFePO4的实测晶体结构为初始构型,运用基于密度泛函理论的CASTEP程序优化体系的原子坐标和晶胞参数.结构弛豫采用BFGS方法,交换相关势的处理分别选用局域密度近似(LDA)下的CA-PZ泛函和广义梯度近似(GGA)下的PBE泛函,同时运用平面波超软赝势方法,截断能取300 eV,布里渊区的k点设置为2×3×4,电子波函数的获取采用Density-mixing方案,轨道占据参数Smearing取0.1 eV.其次,以PBE泛函优化得到的晶体结构为输入几何,在相同理论水平下计算LiFePO4的电子结构,并分析原子电荷与键级分布情况,绘制相关分子轨道图.最后,仍然采用PBE泛函优化得到的晶体结构作为输入几何,运用Morphology模块的BFDH方法计算研究LiFePO4的晶形和晶面参数,精度设置为Ultra-fine.所有计算工作都是在北京理工大学张同来教授课题组完成的.

图1 LiFePO4的晶胞结构Fig.1 Unit cell of LiFePO4
3 结果和讨论
3.1 密度泛函的选择及精度
LiFePO4晶胞结构参数的实验值[5]和计算值列于表1.从表中数据可以看出,由CA-PZ方法计算得到的晶胞参数a、b、c和晶胞体积V均小于相应的实测值,从而导致LiFePO4晶体密度的理论值比实验值大很多,这是LDA泛函的典型特征.然而,PBE泛函优化得到的所有晶胞结构参数与实验值更加接近,并且误差均小于2.5%.由此可见,在计算LiFePO4晶体结构和性质的时候,PBE方法的精度明显高于CA-PZ方法,故本文后续部分均采用PBE方法开展计算研究工作.此外,通过上述分析可知,本文所选用的其它相关计算参数也是准确合理的.
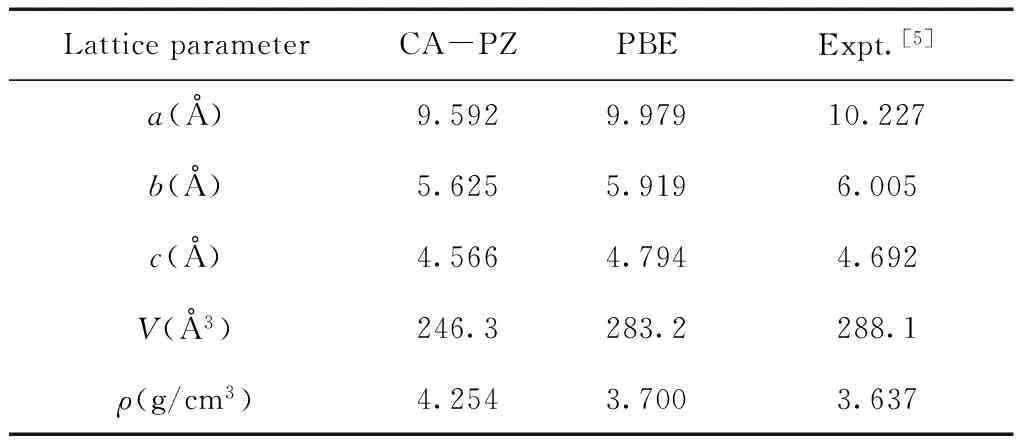
表1 LiFePO4晶胞结构参数的实验值与计算值
3.2 电子结构
在研究原子间键连特性的时候,电荷与重叠布局分析是一种常用的方法[11-13].表2给出了LiFePO4晶体在GGA-PBE理论水平下的Mulliken原子电荷和键级分布情况.从表中数据可以得知,锂离子约带一个单位的正电荷,这表明锂离子与其周围原子之间本质上是离子相互作用,主要靠静电引力结合在一起.然而,铁离子所带的正电荷仅为0.74,和预期的+2价相差甚远,这说明铁离子与配位氧原子之间存在较强的共价相互作用.此外,磷原子约带两个单位的正电荷,而氧原子所带的负电荷介于-1.03 ~ -0.91之间.从表2还可看出,P—O共价键的键级均大于0.6,同时Fe—O配位键也具有较大的键级,且介于0.20 ~ 0.31之间.因此,结合原子的电荷分布情况,可推断Fe—O配位键本质上属于共价键.

表2 LiFePO4的原子电荷与键级分布情况
为了进一步研究Fe—O配位键的成键机制,图2给出了LiFePO4晶体的典型分子轨道图.从图中标注的椭圆形区域可以清晰地看出,铁离子与其邻近的氧原子之间存在较强的价成键电子密度重叠,由此可知,LiFePO4晶体中的Fe—O配位键本质上确实属于共价键,而且该配位键是Fe的3d轨道和O的2p轨道相互靠拢正重叠所形成的[13,14].
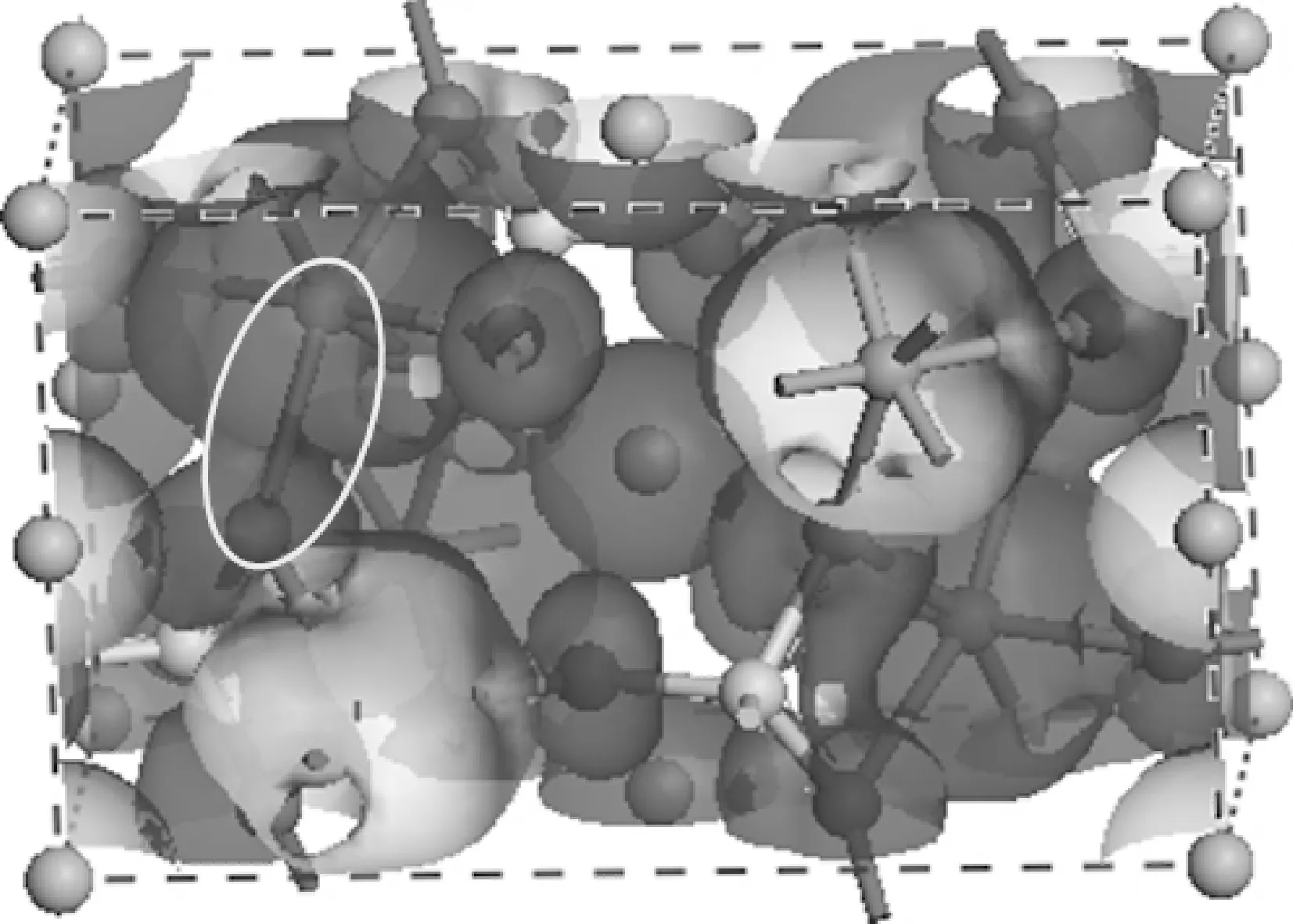
图2 LiFePO4 的典型分子轨道图Fig. 2 The typical molecular orbital picture of LiFePO4
3.3 理论晶形
LiFePO4的实测晶体形貌[7]和利用Morphology模块中的BFDH方法计算得到的理论晶形分别见图3(a)和(b).显然,理论模拟得到的LiFePO4的晶体形貌为两端近似锥形的柱状晶体,长径比等于1.748,这与其块状的实测晶体形貌存在较大差异.通过深入分析可知,这主要是因为理论模拟晶形是LiFePO4在真空中(即自由状态下)的生长晶形,而不是真实环境下的晶形.在实际环境中,溶剂特别是极性溶剂对分子排列方式不同的各晶面造成不同程度和不同方式的影响,进而改变各晶面相对生长速率,导致实际晶形和理论晶形存在显著差异.

图3 LiFePO4的实测晶形(a)和理论晶形(b)Fig. 3 The experimental (a) and calculated (b) crystal morphologies of LiFePO4
LiFePO4理论晶形的特征晶面参数列于表3.从表中数据可以看出,LiFePO4拥有6个特征晶面.值得注意的是,各晶面面积与其百分比在数值上完全相等,这只是巧合而已,并不是所有的物质都会出现这种情况.综合表3和图3的相关信息可知,LiFePO4的理论预测晶形主要是由(1 0 1)、(2 1 0)、(2 0 0)、(0 1 1)、(1 1 1)和(0 2 0)晶面以及它们的对称晶面所构成的.其中,多重度为4的(1 0 1)晶面在晶体中占有最大的比例,其晶面面积为27.4 nm2,占晶体总表面积的27.4%.因此,在LiFePO4晶体的自由生长过程中,(1 0 1)晶面发挥着较大的作用.对于(2 1 0)、(2 0 0)和(0 1 1)晶面来说,它们的晶面面积约为20 nm2.而(1 1 1)和(0 2 0)晶面的面积较小,分别为6.5 nm2和3.3 nm2.

表3 LiFePO4晶体的晶面参数
3.4 晶体生长机理
根据以上分析可知,LiFePO4的理论晶形与其在水溶液中的实际晶形存在很大差异,这主要是溶剂化效应所造成的.那么,溶剂化效应的微观作用机理到底是怎样的呢?通过与LiFePO4的X-射线衍射(XRD)结果[7]进行比较,发现理论预测晶形中晶面面积较小的(0 2 0)晶面在实际晶形中拥有较大的晶面面积.图4给出了LiFePO4晶体中各理论预测晶面的微观结构,即晶面上的原子排列方式.从图4可以看出,各个特征晶面上都有氧原子,但是(0 2 0)晶面的原子占有率相对较低,空间位阻较小,因此溶液中的水分子更容易通过氢键吸附在该晶面上,从而增加了晶面滞流层的厚度,降低了该晶面的相对生长速率,最终使(0 2 0)晶面具有较大的晶面面积,从而在XRD谱图中出现强衍射峰.

图4 LiFePO4各晶面的结构示意图Fig. 4 The crystal facet structures of LiFePO4
此外,实验研究结果表明[7-9],表面活性剂有抑制LiFePO4晶粒长大的作用,这又是为什么呢?众所周知,表面活性剂大多是有机酸或者有机聚合物,故该类体系含有大量的C—H键.如图4所示,LiFePO4的6个特征晶面上都有氧原子,因此表面活性剂和各晶面之间都可以形成大量的C—H…O氢键,从而将LiFePO4晶粒有效地包覆起来,降低其生长速率,最终制备出晶粒细小均匀且形貌规则的LiFePO4正极材料.
4 结 论
采用密度泛函理论对LiFePO4的结构和晶形进行了系统地计算模拟研究,结果表明:
(1)PBE泛函的精度明显高于CA-PZ泛函,因此PBE泛函能够更加准确地预测LiFePO4的结构和性质.
(2)锂离子与其周围原子之间本质上是离子相互作用,而铁离子的3d轨道和氧原子的2p轨道相互重叠形成共价型配位键.
(3)LiFePO4的理论模拟晶形与其实测晶体形貌有较大差异,这主要是因为理论晶形是物质在真空中的生长晶习,而非真实环境下的晶习.
(4)(0 2 0)晶面的原子占有率较低,溶剂分子更易通过氢键吸附在该晶面上,从而增加晶面滞流层的厚度,降低生长速率,使其在实际晶形中拥有较大的面积.
(5)表面活性剂和LiFePO4各晶面之间可以形成大量的C—H…O氢键,将晶粒包覆起来,减小其生长速率,从而抑制LiFePO4晶粒长大.
[1] Padhi A K, Nanjundaswamy K S, Goodenough J B. Phospho-olivines as positive-electrode materials for rechargeable lithium batteries [J].J.Electrochem.Soc., 1997, 144 (4): 1188.
[2] Luo C X, Song H Y, Liao S J. Research progress in preparation and modification of LiFePO4using wet-chemical approaches [J].Mater.Rev., 2010, 24 (4): 34 (in Chinese) [罗传喜, 宋慧宇, 廖世军. 磷酸铁锂的液相合成及改性研究进展 [J]. 材料导报, 2010, 24 (4): 34]
[3] Tang Z Y, Qiu R L, Teng G P,etal. Research progress of LiFePO4cathode material for lithium ion batteries [J].Chem.Ind.Eng.Prog., 2008, 27 (7): 995 (in Chinese) [唐致远, 邱瑞玲, 腾国鹏, 等. 锂离子电池正极材料LiFePO4的研究进展 [J]. 化工进展, 2008, 27 (7): 995]
[4] Zhang K C.ModernCrystallography[M]. Beijing: Science Press, 1987: 58 (in Chinese) [张克从. 近代晶体学基础 [M]. 北京: 科学出版社, 1987: 58]
[5] Garcia-Moreno O, Alvarez-Vega M, Garcia-Alvarado F,etal. Influence of the structure on the electrochemical performance of lithium transition metal phosphates as cathodic materials in rechargeable lithium batteries: a new high-pressure form of LiMPO4(M = Fe and Ni) [J].Chem.Mater., 2001, 13 (5): 1570.
[6] Tang P, Holzwarth N A W. Electronic structure of FePO4, LiFePO4, and related materials [J].Phys.Rev. B, 2003, 68 (16): 165107.
[7] Ellis B, Kan W H, Makahnouk W R M,etal. Synthesis of nanocrystals and morphology control of hydrothermally prepared LiFePO4[J].J.Mater.Chem., 2007, 17 (30): 3248.
[8] Choi D, Kumta P N. Surfactant based sol-gel approach to nanostructured LiFePO4for high rate Li-ion batteries [J].J.Power.Sources, 2007, 163 (2): 1064.
[9] Liang F, Dai Y N, Yao Y C. Effect of surfactant on the electrochemical performance of LiFePO4synthesized by sol-gel method [J].Mater.Rev., 2009, 23 (7): 96 (in Chinese) [梁风, 戴永年, 姚耀春. 表面活性剂对溶胶-凝胶法合成LiFePO4电化学性能的影响[J]. 材料导报, 2009, 23 (7): 96]
[10] Fisher C A J, Islam M S. Surface structures and crystal morphologies of LiFePO4: relevance to electrochemical behaviour [J].J.Mater.Chem., 2008, 18 (11): 1209.
[11] Zhi L L, Li Y Q, Gu L S,etal. Density functional study of the Cd clusters [J].J.At.Mol.Phys., 2012, 29 (1): 76 (in Chinese) [智丽丽, 李艳青, 古丽姗, 等. Cd团簇的第一性原理研究 [J]. 原子与分子物理学报, 2012, 29 (1): 76]
[12] Zhang F C, Li C F, Sun Y A,etal. Density functional theory study on sulfur adsorption on Fe(111) surface [J].J.At.Mol.Phys., 2013, 30 (2): 328 (in Chinese) [张凤春, 李春福, 孙延安, 等. 硫在Fe(111)面吸附的密度泛函研究 [J]. 原子与分子物理学报, 2013, 30 (2): 328]
[13] Zhu Q H, Huang H S, Hu W H,etal. A theoretical study of the structures and properties of Al(N3)3and P(N3)3[J].J.At.Mol.Phys., 2013, 30 (3): 350 (in Chinese) [朱乾华, 黄辉胜, 胡武洪, 等. Al(N3)3和P(N3)3结构与性质的理论研究 [J]. 原子与分子物理学报, 2013, 30 (3): 350]
[14] Wang Q, Li Q, Dai J F,etal. DFT study on the structures and properties of (TiZr)n(n=1~7) clusters [J].J.At.Mol.Phys., 2012, 29 (3): 437 (in Chinese) [王青, 李强, 戴剑锋, 等. (TiZr)n(n=1~7) 团簇的几何构型与性质的密度泛函研究 [J]. 原子与分子物理学报, 2012, 29 (3): 437]
Theoretical study of the electronic structure and crystal morphology of LiFePO4as cathode material for lithium battery
HUANG Hui-Sheng, HU Wu-Hong, ZHANG Guo-Qing, ZHANG Fu-Lan, DING Shi-Min
(1.Chongqing Key Laboratory of Inorganic Special Functional Materials, Chongqing 408100, China; 2.School of Chemistry and Chemical Engineering, Yangtze Normal University, Chongqing 408100, China)
The geometric and electronic structures, crystal morphology, crystal facet area and the crystal facet relative growing velocity of LiFePO4were theoretically investigated by the CASTEP and Morphology modules. The obtained results show that the PBE functional has better accuracy than CA-PZ functional. The Fe—O coordination bonds are covalent in nature. Moreover, the simulated crystal morphology is columnar, which is obviously different from the experimental crystal morphology of flake. This is due to the calculated crystal morphology is the crystal habit of LiFePO4under the vacuum condition, not in the actual environment. For the experimental morphology of LiFePO4, the area of the (0 2 0) crystal facet is large, which is closely related with the atomic arrangement and density on the crystal surface. Additionally, there are many C—H…O hydrogen bonds between surfactant and crystal facets of LiFePO4, the crystallites are coated and their growing velocities slow down, and so the surfactant can control crystallite growth.
LiFePO4; Electronic structure; Crystal morphology; Solvent effect; Surfactant
2014-08-15
重庆市自然科学基金(cstc2011jjA50013);重庆市教委科技计划项目(KJ111310)
黄辉胜(1982—),男,重庆人,副教授,主要从事高能量密度材料研究.E-mail: huanghuisheng508@126.com
103969/j.issn.1000-0364.2015.10.025
O641
A
1000-0364(2015)05-0870-05
猜你喜欢
中学生数理化(高中版.高考理化)(2019年11期)2019-11-30
中学生数理化(高中版.高考理化)(2019年11期)2019-11-27
中学生数理化(高中版.高考理化)(2019年10期)2019-11-08
物理实验(2019年7期)2019-08-06
中学化学(2019年1期)2019-06-29
航空材料学报(2019年2期)2019-04-15
西安工业大学学报(2018年6期)2018-02-13
中国造纸(2015年7期)2015-12-16
云南民族大学学报(自然科学版)(2015年4期)2015-11-14
中国洗涤用品工业(2015年2期)2015-02-28
