
量子化学方法研究噻吩与过渡金属M=(Mo,Pd,Sn)的吸附行为
2015-03-17 11:58龙威
黑龙江大学工程学报 2015年3期
龙 威
(南华大学 化学化工学院,湖南 衡阳 421001)
量子化学方法研究噻吩与过渡金属M=(Mo,Pd,Sn)的吸附行为
龙 威
(南华大学 化学化工学院,湖南 衡阳 421001)
基于已有的实验基础上,利用量子化学方法结合Genecp基组水平上研究了噻吩分子与3种过渡金属M= (Mo,Pd,Sn)的吸附微观行为。计算结果表明:不同的过渡金属原子对噻吩分子的吸附存在着不同的吸附位,过渡金属Mo吸附存在多种吸附位以β、θ位为主,且吸附后能量分别降低了328.795kJ/mol和327.868kJ/mol;过渡金属Pd吸附以δ位为主,吸附后能量降低了380.654kJ/mol;过渡金属Sn吸附以α、δ位为主,吸附后能量分别降低了272.514和512.130kJ/mol。吸附能量的计算应考虑零点能的校正,B3LYP方法在构型优化和能量计算上均具有较高的精度和优势。
噻吩裂解;过渡金属;量子化学;吸附行为
0 引 言
石油作为传统的化石能源,在全球工业上具有不可代替的地位[1]。石油的加工一直是科学研究的热点和难点,而清洁能源与环境保护越发受到全球人类的关注[2],因而,如何深度加工化石能源,使它不会对环境造成污染是人类急需追求的化学工艺目标[3]。工业革命时期,人类就已经大量利用化石能源导致过多的SO2气体排放到大气而形成酸雨,严重地危害了地球上生物的生存,追溯其根源来自于含煤化合物的未妥善处理[4]。含硫的化合物种类繁多,其脱硫工艺复杂无法统一化,导致含硫化合物的脱硫工艺一直未成熟,而环境保护、持续发展的人类生存要求需要重点研究含硫有机物的催化转化过程[5]。
有机含硫化合物的脱硫是一个难题[6],许多有机化合物中含有S原子的环状结构,其分子非常稳定,环内共轭效应十分强烈,常规的脱硫方法发挥不了作用。噻吩,作为有机含硫化合物的典型代表,在有机含硫化合物中占有很大的比例,它的分子又具有非常稳定的环状结构,其脱硫过程复杂。最近,过渡金属具有良好的催化性能引起了科学界的关注,通过过渡金属催化噻吩分子的脱硫,达到了良好的效果[7]。
早在2008年,Yu等[8]通过实验比较了Co/Ni/Mo负载在碳纳米管上对噻吩的催化性能,指出过渡金属Mo能发挥最佳活性;最近,Eduardo等[9]通过实验表明过渡金属Mo掺杂在Ni中能发挥很强的催化噻吩脱硫的效果,指出过渡金属Mo是噻吩脱硫反应最佳的催化金属;而Zdeněk等[10]则通过实验指出过渡金属Pd或Pd-Pt能对噻吩发挥良好的催化效应;Valeria等则提出过渡金属Pd-Au催化下加氢能较好地促进噻吩的脱硫;另外,Biswajit等[11]通过实验发现了过渡金属Sn对噻吩的脱硫也可发挥很强的催化活性;我国的Zhang等[12]也探究了ZnO作为载体上Mo,Pd,Sn等过渡金属均对噻吩的高效催化脱硫过程,提出了它们对噻吩的脱硫均具有较强的催化活性。其他过渡金属催化噻吩的脱硫过程,许多文献[13-14]虽有报道,但其催化活性参差不齐,无法形成统一的脱硫机理。
通过总结过渡金属在噻吩的催化脱硫过程的实验研究[15],发现金属M=(Mo,Pd,Sn)相对其他过渡金属具有理想的催化活性。由于噻吩自身有毒,且过渡金属价格昂贵,实验研究噻吩的催化效应具有较大的难度,笔者选择量子化学方法对过渡金属M=(Mo,Pd,Sn)与噻吩分子的吸附行为进行了研究。在此方面,虽也有相关报道,如郑柯文等[16]用量子化学方法提出了噻吩容易在分子筛上形成带正碳离子的中间体,进而与烯烃加成发生脱硫反应生成己烯;徐坤等[17]利用密度泛函理论提出了噻吩在γ-Mo2N(100)表面上加氢脱硫反应的机理,但这些理论研究比较片面且有所不足。基于笔者在这方面的研究基础和经验[18-19],选择量子化学研究方法进行探究,通过计算探索3种不同的过渡金属原子对噻吩分子的具体吸附行为,可为进一步实验做科学合理的指引,具有重要的研究意义。
1 计算方法
首先采用密度泛函理论(Density Functional Theory,DFT)中的B3LYP方法对噻吩与过渡金属M= (Mo,Sn,Pd)原子的吸附模型进行了研究,然后利用HF、MP2、CCSD等量化方法进行能量精度的验证计算。对于过渡金属原子,采用赝势基组lanl2dz,其它原子选择6-311+G(d,p)基组,计算过程中使用genecp方法将基组拟合。使用B3LYP方法对各物种进行了几何构型优化及频率分析,发现所有的振动频率均为正值,说明吸附前后的各物种是势能面上的极小点。为了能量的精确度,在频率分析的同时进行了零点能的校正,其校正因子取0.97,获得对应的不同的相对的能量大小数据,吸附的能量ΔEabs= (E产物+Ezpe) - (E反应物+Ezpe),其中E产物为发生吸附后形成稳定中间体的能量,E反应物为发生吸附前噻吩分子和过渡金属原子的能量之和,Ezpe为零点能校正数值,ΔE为考虑零点能校正后的相对能量,以上全部工作用Gaussian 03程序[20]在南华大学的计算化学微型计算机上完成。
2 结果与讨论
2.1 吸附行为模型
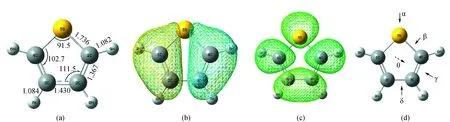
图1 噻吩分子的几何结构、分子轨道、吸附模式图Fig.1 Geometric parameters,molecular orbital and adsorption model of thiophene molecule
Table 1 The atomic Mulliken charge distribution of thiophene molecule

原子Mulliken电荷原子Mulliken电荷1C-0.40326H0.18702C-0.18147H0.17003C-0.13738H0.15144C-0.37659H0.19395S0.3961
2.2 过渡金属Mo原子与噻吩的吸附行为

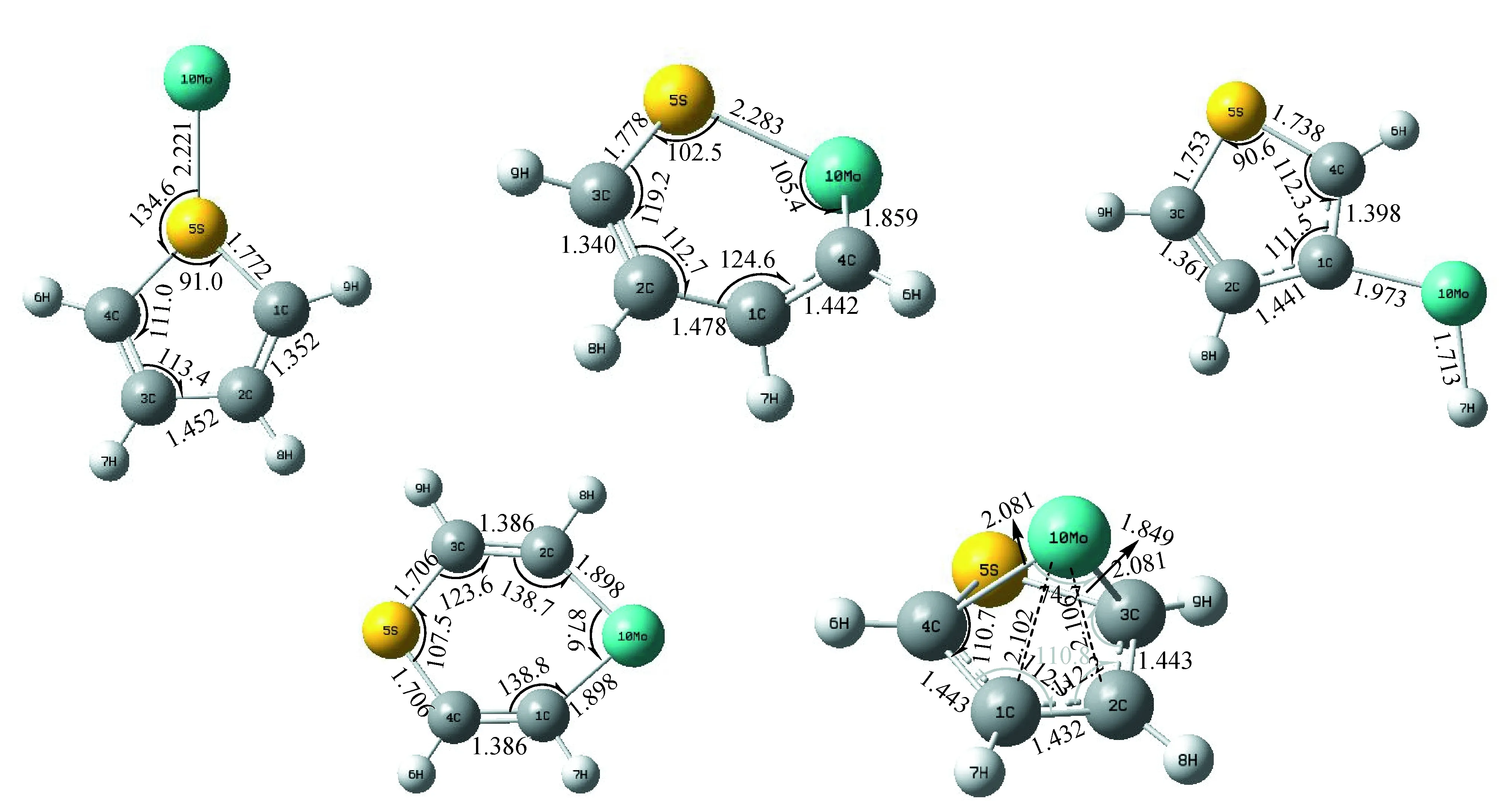
图2 过渡金属Mo与噻吩吸附的5种模式Fig.2 Five adsorb patterns between Mo and thiophene molecule
考虑了这5种种吸附位下的吸附能大小,将其能量数据列于表2。不难发现,吸附后产物的能量均比反应物的能量低,其中ΔEabs为未考虑零点能校正的吸附能数据,ΔE’abs为考虑零点能校正后的吸附能数据,而ΔE为两种模式下相对的能量大小。吸附能负值越大,表明吸附后整个体系越稳定。比较数值可知,B3LPY方法计算的过渡金属Mo原子在β、θ位吸附噻吩分子后形成的体系较为稳定,其能量比α位吸附位分别低328.795和327.868 kJ/mol,表明过渡金属Mo比较适合在这两个方位吸附噻吩并发生催化裂解反应。为了进一步衡量能量的精度,以同样的几何构型在相同的基组水平上利用HF、MP2、CCSD等方法进行计算,相应的ΔE也列于表2中,与B3LYP方法计算的趋势基本一致。
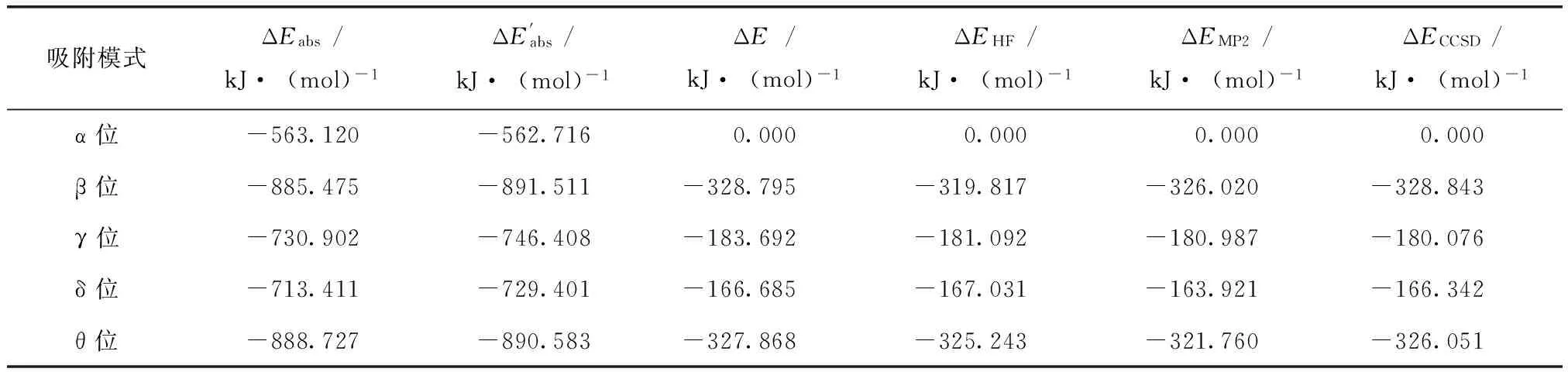
表2 过渡金属Mo与噻吩吸附的能量数据
2.3 过渡金属Pd原子与噻吩的吸附行为
计算研究表明,过渡金属Pd原子对噻吩分子的吸附不同于Mo,笔者找到了4种模式,即β、γ、δ、θ位,但其稳定的吸附只有δ位一种。过渡金属Pd原子最外层电子排布是4d10,d轨道上电子已呈现排满状态,故过渡金属Pd原子反应活性较低。图3的几何结构表明:过渡金属Pd原子从β、γ及θ位吸附噻吩分子时,基本不会影响噻吩环的分子结构,而δ位发生吸附时则破坏了噻吩分子的环状结构,C-C键被过渡金属Pd原子打断而形成六元环的平面结构;同样,当发生β位和θ位吸附时,过渡金属原子偏向了一边,在分子环平面的上方或下方主要靠近了连接S原子的C原子且与之成键,这种不规则且不稳定的吸附主要是由于最外层没有容易给出的单电子的缘故导致。相关的吸附能数据见表3,发现仅有δ位发生吸附时能量有较大的降低,说明它是稳定的吸附模式,吸附后能量降低了380.654 kJ/mol,而其他位发生吸附后能量反而有所增加,这可能是S原子的孤对电子对与过渡金属的4d半充满电子轨道相抵制的缘故。为了进一步衡量能量的精度,以同样的几何构型在相同的基组水平上利用HF、MP2、CCSD等方法进行计算,获得的ΔE也列于表3中,与B3LYP方法计算的趋势基本一致,提高了计算的可信度。

图3 过渡金属Pd与噻吩吸附的4种模式图Fig.3 Four adsorb patterns between Pd and thiophene molecule

吸附模式ΔEabs/kJ·(mol)-1ΔE'abs/kJ·(mol)-1ΔE/kJ·(mol)-1ΔEHF/kJ·(mol)-1ΔEMP2/kJ·(mol)-1ΔECCSD/kJ·(mol)-1β位967.926969.695683.685684.029690.331683.076γ位312.105313.04227.03226.98725.03127.009δ位-373.754-380.654-666.664-659.098-661.703-666.523θ位285.062286.0100.0000.0000.0000.000
2.4 过渡金属Sn原子与噻吩的吸附行为

图4 过渡金属Sn与噻吩吸附的4种模式图Fig.4 Four adsorb patterns between Sn and thiophene molecule

吸附模式ΔEabs/kJ·(mol)-1ΔE'abs/kJ·(mol)-1ΔE/kJ·(mol)-1ΔEHF/kJ·(mol)-1ΔEMP2/kJ·(mol)-1ΔECCSD/kJ·(mol)-1α位-272.952-272.514-1018.500-1016.323-1015.684-1018.492β位1836.9021835.3161089.3301086.4651081.0361089.292δ位-507.488-512.130-1258.116-1257.045-1255.340-1258.099θ位747.054745.9860.0000.0000.0000.000
3 结 论
通过对3种过渡金属M= (Mo,Pd,Sn)对噻吩分子吸附模型的量化计算研究,发现了不同的过渡金属存在不同的吸附模式,通过比较各个吸附模式的能量数据大小关系,找到了不同金属吸附的最佳稳定吸附位,对反应机理的进一步研究具有积极的促进意义:
1)过渡金属Mo原子有5种吸附位,且每种吸附发生后体系能量均有所下降,但过渡金属Pd、Sn仅存在有少量吸附位,能量数据表明它们分别只有一种和两种稳定的吸附模式。
2)过渡金属Mo原子吸附噻吩分子的位置以β、θ位为主,其吸附后能量分别降低了328.795和327.868 kJ/mol;过渡金属 Pd原子吸附噻吩分子的位置以δ位为主,其吸附后能量降低了380.654 kJ/mol;过渡金属Sn吸附噻吩分子的位置以α、δ位为主,其吸附后能量分别降低了272.514和512.130 kJ/mol,吸附后体系能量降低才为稳定的吸附行为。
3)过渡金属Mo原子对噻吩吸附位较多,表明它与噻吩分子反应活性更大,能以不同的方位吸附噻吩分子进而发生催化开环脱硫,这与相关实验文献[9,12]报道相一致。
4)吸附能计算时应考虑零点能校正的因素影响,未考虑零点能校正的吸附能量偏差较大,这是在量化计算中能量叠加中着重考虑且应消除的误差。相对其他方法,B3LYP方法在构型优化和能量计算上均具有较高的精度和优势,CCSD方法与B3LYP方法计算的能量数据相近。
[1]Baeza P,Aguila G,Vargas G,et al.Adsorption of thiophene and dibenzothiophene on highly dispersed Cu/ZrO2absorbents[J].Applied Catalysis B:Environmental,2012,111-112:133-140.
[2]Saha B,Sengupta S.Influence of different hydrocarbon components in fuel on the oxidative desulfurisation of thiophene:Deactivation of catalyst[J].Fuel,2015,150(15):679-686.
[3]Potapenko O,Doronin V P,Sorokina T P,et al.Transformations of thiophene compounds under catalytic cracking conditions[J].Applied Catalysis B:Environmental,2012:117-118.
[4]Bezverkhyy L,Ryzhikov A,Gadacz G,et al.Kinetics of thiophene reactive adsorption on Ni/SiO2and Ni/ZnO[J].Catalysis Today,2008,130(1):199-205.
[5]Dong Kunming,Ma Xiaoming,Zhang Hongbin,et al.Novel MWCNT-Support for Co-Mo Sulfide Catalyst in HDS of Thiophene and HDN of Pyrrole.Journal of Natural Gas Chemistry,2006,15(1):28-37.
[6]Pawelec B,Mariscal R,Navarro R M,et al.Simultaneous 1-pentene hydroisomerisation and thiophene hydrodesulphurisation over sulphided Ni/FAU and Ni/ZSM-5 catalysts[J].Applied Catalysts A:GENERAL,2004,262(2):155-166.
[7]Oleg V P,Vladimir P D,Tatyana P S,et al.Transformations of thiophene compounds under catalytic cracking conditions[J].Applied Catalysis B:Environmental,2012,117-118(3):177-184.
[8]Yu Zhixin,Lars Erik F,Kjell M,et al.Hydrodesulfuriza tion of thiophene on carbon nanofiber supported Co/Ni/Mo catalysts[J].Applied Catalysis B: Environment al,2008,84(3-4):482-489.
[9]Eduardo P B,Alexandre B F ,Alano V S N,et al.Incorporation of the precursors of Mo and Ni oxides directly into the reaction mixture of sol-gel prepared γ-Al2O3-ZrO2supports-Evaluation of the sulfided catalysts in the thiophene hydrodesulfuriza tion[J].Catalysis Today,2015,246(3):184-190.
[10]ZdeněkV,HanaK,LuděkK,etal.EffectofpreparationofPdandPd-Ptcatalystsfromacidleachedsilica-aluminaontheiractivityinHDSofthiopheneandbenzothiophene[J].AppliedCatalysisB:Environmental,2011,108-109(10):152-160.
[11]BiswajitS,SonaliS.Influenceofdifferenthydrocarboncomponentsinfuelontheoxidativedesulfurisationofthiophene:Deactivationofcatalyst[J].Fuel,2015,150(6):679-686.
[12]ZhangJingcheng,LiuYunqi,TianShuang,etal.ReactiveadsorptionofthiopheneonNi/ZnOadsorbent:EffectofZnOtexturalstructureonthedesulfurizationactivity[J].JournalofNaturalGasChemistry,2010,19(3):327-332.
[13]ValeriaLP,MariaLT,AnnaMV.PdandPdAucatalystssupportedover3-MPTESgraftedHMSusedintheHDSofthiophene[J].AppliedCatalysisB:Environmental,2012,119-120(5):248-255.
[14]JoseN,SenguptaS,BasuJK.OptimizationofoxidativedesulfurizationofthiopheneusingCu/titaniumsilicate-1bybox-behnkendesigen[J].Fuel,2011,90(2):626-632.
[15]龙 威,颜雪明.过渡金属在石油脱硫技术中的催化作用[J].佛山科学技术学院学报:自然科学版,2013,31 (4):22-29.
[16]郑柯文,高金森,徐春明.噻吩催化劣化脱硫机理的量子化学分析[J].化工学报,2004,55(1):87-90.
[17]徐 坤,冯 杰,褚 绮,等.噻吩在γ-Mo2N(100)表面上加氢脱硫反应的密度泛函理论研究[J].物理化学学报,2014,30(11):2 063-2 070.
[18]徐文媛,龙 威,杜瑞焕.镍基上CH4脱氢与超临界CO2重整的量化计算[J].化学通报,2011,74(8):732-736.
[19]龙 威,颜雪明,陈仲清.BiOX(X=F,Cl,Br,I)的溶剂效应、电子结构和光学性质[J].黑龙江大学自然科学学报,2013,30(5):635-641.
[20]MalickDK,PeterssonGA,MontgomeryJrJA.TransitionstatesforchemicalreactionsI:Geometryandclassicalbarrierheight[J].TheJournalofChemicalPhysics,1998,108(104):5 704-5 713.
[21]张连阳,施 炜,夏盛杰,等.Au/Pd(111)双金属表面催化噻吩加氢脱硫的反应机理[J].物理化学学报,2014,30(10):1 847-1 854.
Adsorption behavior between thiophene and M=(Mo,Pd,Sn) by quantum chemistry method
LONG Wei
(School of Chemistry and Chemical Engineering,University of South China,Hengyang 421001,Hunan,China)
Based on the existing experiment,Gaussian 03 package to study the adsorption of microscopic behavior between thiophene molecule and three transition metals as M=(Mo,Pd,Sn) were used,which combine with the quantum chemistry method and the genecp basis set.It is showed that there are many different molecular adsorb patterns between the different transition metal atoms and thiophene.The transition metal Mo is given more priority to occur the β and θ adsorbing model,and the decreased energy was 328.795 and 327.868 kJ/mol respectively,transition metal Pd is given more priority to occur the δ adsorbing model,and the decreased energy as high as 380.654 kJ/mol;Transition metal Sn is given more priority to occur the α and δ adsorbing model,and the decreased energy was 272.514 and 512.130 kJ/mol respectively.The calculation of adsorption energy should consider the zero-point energy correction.Compared with other methods,B3LYP method is more advantage about optimization and energy calculation.
thiophene cracking;transition metal;quantum chemistry;adsorption behavior
10.13524/j.2095-008x.2015.03.044
2015-06-15
衡阳市科技局基础研究资助项目(2013KJ23)
龙 威(1983-),男,湖南湘潭人,实验师,博士研究生,研究方向:生物质加氢工艺和反应催化机理研究,E-mail:usclw2013@yeah.net。
O641
A
2095-008X(2015)03-0039-07
猜你喜欢
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
国学(2020年1期)2020-06-29
摄影之友(影像视觉)(2017年10期)2017-11-07
摄影之友(影像视觉)(2017年1期)2017-07-18
当代化工研究(2016年1期)2016-03-16
合成化学(2015年10期)2016-01-17
应用化工(2014年9期)2014-08-10
无机化学学报(2014年9期)2014-02-28
