
吗替麦考酚酸酯的合成工艺优化
2014-12-14 07:08胥秀英张心蕊陈海芹
重庆理工大学学报(自然科学) 2014年1期
刘 红,胥秀英,任 杰,张心蕊,陈海芹,王 锐
(重庆理工大学药学与生物工程学院,重庆 400054)
吗替麦考酚酯(mycophenolate mofetil,MPM)是由瑞士罗氏(Roche)公司研发的一种新型免疫抑制剂,于1995年首次在美国上市,商品名为骁悉,在体内脱酯化后形成具有免疫抑制活性的代谢产物麦考酚酸(mycophenolic acid,MPA)。麦考酚酸酯最初是作为一种抗细菌和抗真菌的药物,20世纪60年代后期开始作为抗肿瘤药物应用于临床。直到20世纪80年代,在寻找高选择低毒性的免疫抑制剂治疗自身免疫性疾病的药物时,麦考酚酸酯的抑制免疫作用才被学术界发现。1995年5月,麦考酚酸酯获得美国食品药品管理局(FDA)批准用于预防肾移植急性排异反应[1-3]。其免疫抑制的作用机制是选择性抑制与排斥反应相关的T淋巴细胞和B淋巴细胞,并且有抑制动脉平滑肌增生的作用,后者是目前其他免疫抑制剂所不具备的[4]。
由于吗替麦考酚酯优秀的药用价值,其合成方法广受关注。专利US 4753935[5]揭示了2种不同的制备路线:第1种路线为由MPA制备成相应酰氯,再完成缩合。此路线缺点在于需要两步合成步骤,酰氯会对设备产生腐蚀,反应过程中产生大量的二聚杂质。另外,由于痕量铁的污染,产物存在严重的颜色问题。第2种路线中羧酸经碳亚胺活化后再缩合,此路线由于碳亚胺类活化剂的成本较高,使其难以工业化,终产物酯的纯度亦较低。专利 US 5247083[6]制备方案为MPA在有机溶剂中经恒沸除水直接酯化,所用溶剂为甲苯、间二甲苯、二氯甲烷或其混合物,此路线的局限在于反应时间较长(60~100 h),得到紫色麦考酚酯晶体,产物纯度低。WO 2004089946[7]以微波激发合成MPM,此法成本昂贵,难以实现工业化应用。专利 WO 2003042393[8]、WO 2000034503[9]、WO 2006024582[10]利用生物酶催化合成 MPM,缺点是反应时间较长、收率较低、有大量反应原料残留。专利WO 02100855[11]利用高沸点溶剂直接酯化,值得关注的是实验证实二丁醚极其有效,但产物颜色问题仍有改进空间。
迄今为止,全球吗替麦考酚酯合成最主要的问题在于产物纯度低,常常显紫色,反应时间长,收率低。因此,为了解决麦考酚酯工业化生成中的上述诸多问题,获得高纯度、高收率的麦考酚酸酯,急需对麦考酚酯现有的制备方法进行系统优化,以获得最佳工艺条件。
1 反应溶剂优化
1.1 实验方法
氮气保护下,10 g麦考酚酸与20 mL溶剂混合,剧烈搅拌并升温至50~60℃,加入5 mL 2-羟乙基吗啉,恒沸除水条件下反应(TLC监控),冷至室温并加入50 mL二氯甲烷。溶液以1%稀氨水(20 mL×2)、水(50 mL)洗涤。真空旋除溶剂,并以乙酸乙酯结晶,真空干燥,可得产品MPM。
1.2 实验结果
由表1的实验数据可知:选用溶剂丁醚的实验结果最佳;均三甲苯虽收率相当,但反应温度相对较高;甲苯、间二甲苯、邻二甲苯获得较次的结果。即:10 g MPA和5 mL羟乙基吗啉在20 mL丁醚的条件,在145℃下反应15 h可高收率获得MPM。
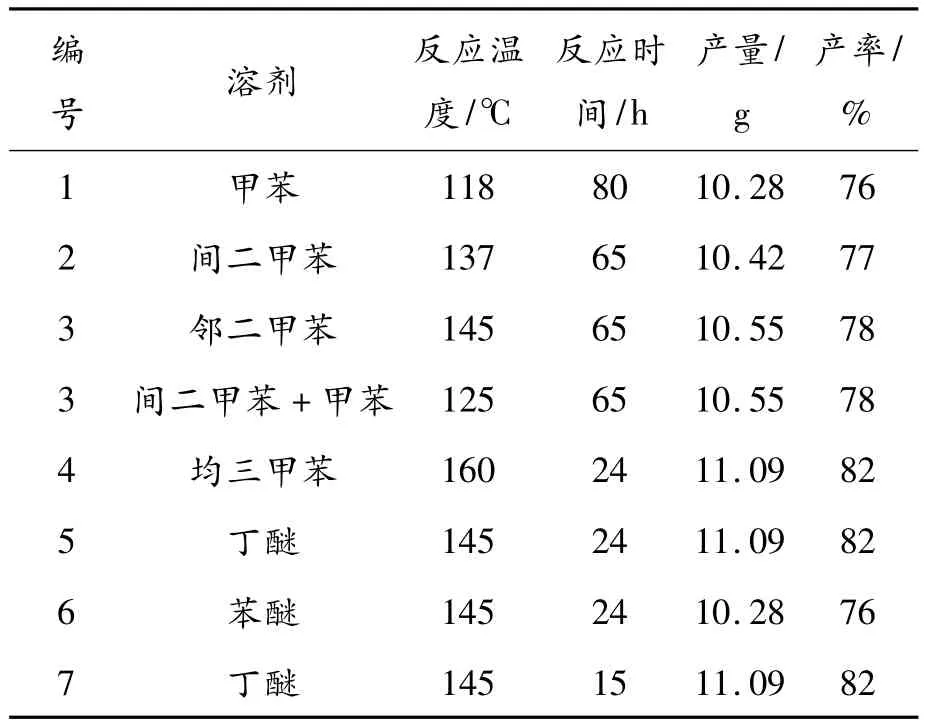
表1 麦考酚酸酯合成工艺之反应溶剂优化
1.3 实验结论
二丁醚为最佳反应溶剂,但产物的颜色仍有改进空间,即纯化过程有待进一步改进。
2 混合溶剂结晶优化
结晶有助于除去欧洲药典所定义的杂质,如麦考酚酸酯的内酯。溶剂优选为混合溶剂,如甲醇、乙醇、丙醇或异丙醇与乙酸乙酯的混合溶剂。结晶应在合适的温度下进行,加热溶解后,加入螯合剂脱色,在残留物紫色消失后趁热过滤,冷置导致晶体析出。
2.1 实验方法
氮气保护下,50 g麦考酚酸与100 mL丁醚混合,剧烈搅拌并升温至50~60℃,加入27 mL 2-羟乙基吗啉,恒沸除水条件下反应15 h后,冷至室温并加入250 mL二氯乙烷。溶液以1% 稀氨水(100 mL×2)、水(250 mL)洗涤。真空旋除溶剂,得粗产物MPM。残留物按照表2内容分批结晶。
氮气保护下将5 g左右残留物(粗MPM)溶解在混合溶剂中(约40 mL)升温至50~60℃。结晶方法如下:取5克样品,加入适量易溶溶剂(如丙醇),在加热状态下(60℃)搅拌使其全溶,然后缓慢加入难溶溶剂((如乙酸乙酯)至其刚好浑浊,随后加入少量易溶溶剂使其刚好澄清,放置,缓慢冷却至0~5℃,并搅拌1 h。过滤晶体,并以冷的醇(0~5℃,10 mL)洗涤。真空干燥,可得纯产品MPM。
2.2 实验结果
由表2的实验数据可知,混合溶剂比例丙醇∶乙酸乙酯=5∶1为最佳,在该比例条件下获得纯品MPM的产率是最高的,即该比例为最佳的结晶比例。甲醇、乙醇为结晶溶剂时,产物溶解性增强,须多次结晶。

表2 麦考酚酸酯合成工艺的混合溶剂比例优化
2.3 实验结论
以丙醇∶乙酸乙酯=5∶1为混合溶剂结晶,强于乙酸乙酯及其他溶剂组合,为MPM最佳结晶溶剂。但是在该比例条件下产物颜色问题仍有提高空间,故现行的实验方法还需进一步优化。
3 铵盐类螯合剂优化
3.1 实验方法
氮气保护下,10 g麦考酚酸与20 mL正丁醚混合,剧烈搅拌并升温至50~60℃,加入5 mL 2-羟乙基吗啉,恒沸除水条件下反应(15 h,TLC监控),减压蒸出有机溶剂,残留固体冷至室温并加入50 mL二氯甲烷。溶液以1%稀氨水(20 mL×2)、水(50 mL)洗涤。真空旋除溶剂。氮气保护下,将残留物溶解在丙醇/乙酸乙酯(75 mL∶15 mL)的混合溶剂中升温50~60℃。随后,将螯合剂(1 mmol/2 mL热蒸馏水)加入,当残留物紫色消失后,趁热过滤,固体用16 mL丙醇/乙酸乙酯混合液(50~60℃)洗涤,合并滤液,缓慢冷却至0~5℃,并搅拌1 h。过滤晶体,并以20 mL丙醇(0~5℃)洗涤,真空干燥,可得产物。
3.2 实验结果
由表3的实验数据可知,加入螯合剂四甲基乙二胺和乙二胺四乙酸四钠的效果较好,结晶产率较高。
3.3 实验结论
加入螯合剂达到了解决产物颜色和纯度(如表3)的目的,但是在结晶后产率相对较低。因此,继续筛选一些其他的螯合剂,希望能尽量使产率提高。
4 离子液体类螯合剂优化
离子液体由于其可调的结构,独特的特性(环境友好、低蒸汽压、高热稳定性、高化学稳定性、离子迁移及扩散速度快等),其在合成、催化、分离技术、电化学、分析化学以及纳米技术领域得到广泛应用。离子液体在溶剂萃取、废旧高分子化合物的回收、燃料电池和太阳能电池、工业废气中二氧化碳的提取、地质样品的溶解、核燃料和核废料的分离与处理等方面也显示出潜在的应用前景[12-18]。离子液体作为螯合剂纯化物质的能力方面的研究目前尚未有报道,对离子液体进行系列筛选,考察其螯合能力,意义深远。
4.1 实验方法
根据本文实验方法制备粗产物MPM。氮气保护下将10 g左右粗产物溶解在丙醇/乙酸乙酯(68.75 mL∶13.75 mL)的混合溶剂中(丙醇∶乙酸乙酯=5∶1,升温至50~60℃)。随后,将螯合剂(10 mg/1 mL热蒸馏水)加入,当残留物紫色消失后,趁热过滤,固体以热的丙醇/乙酸乙酯混合液洗涤(16 mL),合并滤液,缓慢冷却至0~5℃,并搅拌1 h。过滤晶体,并以冷的丙醇(0~5℃,20 mL)洗涤。真空干燥,可得高纯产品MPM。
4.2 实验结果
由表4的实验数据可知,加入离子液体螯合剂进行结晶纯化的结晶产率均较高,且含量也很理想。其中以离子液体螯合剂1-乙基-3-甲基咪唑溴盐的效果最佳,产率最高为79%。
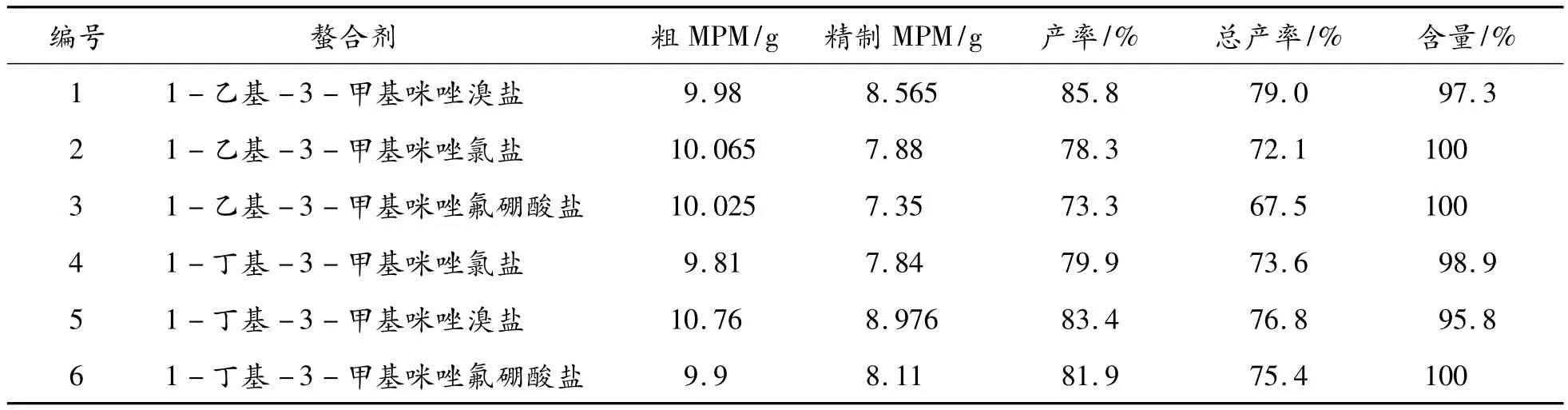
表4 麦考酚酸酯合成工艺的离子液体类螯合剂优化
4.3 实验结论
通过以上对麦考酚酯的合成工艺优化的实验,基本上能得到纯度高、颜色白且产率较高的纯MPM。但是考虑到该工艺要在工业上实现,有必要对合成工艺的成本进行评估,尽量在得到纯度、颜色和产率均较好的纯MPM的条件下使成本较低。笔者认为,在成本估算中,螯合剂的影响较为突出,因此,在不影响纯MPM的质量的前提下,应尽量减少螯合剂的用量,以减少成本。
5 螯合剂用量的优化
5.1 实验方法
氮气保护下,麦考酚酸10 g与20 mL二丁醚混合,剧烈搅拌并升温至50~60℃,加入5 mL 2-羟乙基吗啉,恒沸除水条件下反应15 h后,减压蒸出有机溶剂,残留固体加入80 mL乙酸乙酯。溶液用20 mL饱和碳酸氢钠洗涤2次、50 mL自来水洗涤2次,减压蒸除有机溶剂。氮气保护下将残留物溶解在丙醇/乙酸乙酯(75 mL∶15 mL)的混合溶剂中(丙醇/乙酸乙酯 =5∶1,升温 50~60℃)。随后,将1-乙基-3-甲基咪唑溴盐(溶于热蒸馏水)加入,当残留物紫色消失后,趁热过滤,固体用16.5 mL丙醇/乙酸乙酯混合液(50~60℃)洗涤,合并滤液,加入吗替麦考酚酯纯品0.05 g引晶,缓慢冷却至0~5℃,并搅拌1 h。过滤晶体,并以25 mL丙醇(0~5℃)洗涤,真空干燥,可得白色MPM晶体。
5.2 实验结果
根据该实验方法,加入不同量的1-乙基-3-甲基咪唑溴盐离子液体螯合剂进行结晶纯化粗产品MPM,实验中得到的相应的数据及结果见表5。

表5 麦考酚酸酯合成工艺之1-乙基-3-甲基咪唑溴盐用量优化
由表5的实验数据可知:当螯合剂1-乙基-3-甲基咪唑溴盐的量与MPA的量比例在0.1%(摩尔百分比)时,可以达到纯化的目的,高纯度获得产物。因此要达到不影响MPM质量的目的,螯合剂1-乙基-3-甲基咪唑溴盐的量至少要在0.1%(摩尔百分比)以上。
6 晶种量的优化
在以上筛选出的最佳工艺条件下,通过在纯化结晶步骤中加入少量晶种引晶可提高产率。为了确定所加晶种的量,设计不同组实验进行筛选优化,以期在提高产率的条件下加晶种的量最少。
6.1 实验方法
根据本文实验方法制备粗产物MPM。氮气保护下将10 g左右粗产物溶解在丙醇∶乙酸乙酯(68.75 mL∶13.75 mL)的混合溶剂中(丙醇∶乙酸乙酯=5∶1,升温50~60℃)。随后,将离子液体螯合剂1-乙基-3-甲基咪唑溴盐(5 mg/1 mL热蒸馏水)加入,当残留物紫色消失后,趁热过滤,固体以热的丙醇/乙酸乙酯混合液洗涤(16 mL),合并滤液,缓慢冷却至0~5℃,加入一定量的纯MPM晶种并搅拌1 h。过滤晶体,并以冷的丙醇(0~5℃,20 mL)洗涤。真空干燥,可得高纯产品MPM。
6.2 实验结果
由表6的实验数据可知:当所加晶种的量为0.5% ~0.7%/g(粗产物)为最佳,因为这个比例的加量既能使产率最大量提高,又能使所加晶种的量不是很大。
7 结束语
通过优化实验研究,优化出最佳合成工艺:氮气保护下,麦考酚酸和稍过量2-羟乙基吗啉在以丁醚为反应溶剂下反应,得到粗产品麦考酚酸酯;然后用丙醇∶乙酸乙酯(5∶1)的混合溶剂加热溶解,用0.1%(摩尔百分比)(基于MPA物质量)的1-乙基-3-甲基咪唑溴盐离子液体螯合剂脱色,用含粗品MPM量的0.5% ~0.7%的纯MPM晶种进行结晶,可高纯度(>99%)、高收率(84%)获得产品麦考酚酸酯。
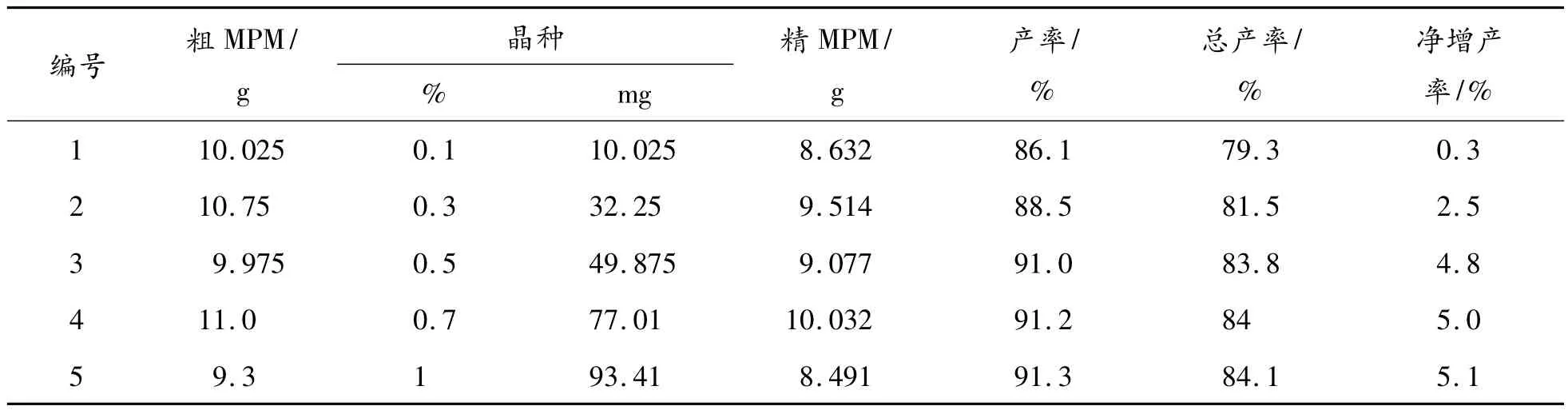
表6 麦考酚酸酯合成工艺的晶种用量优化
代表性合成工艺如下:氮气保护下,麦考酚酸20 g及40 mL二丁醚混合,剧烈搅拌并升温至50~60℃,加入10 mL 2-羟乙基吗啉,恒沸除水条件下反应15 h后,减压蒸出有机溶剂,残留固体加入150 mL乙酸乙酯。溶液用20mL饱和碳酸氢钠洗涤2次、50 mL自来水洗涤2次,减压蒸除有机溶剂。氮气保护下将残留物溶解在丙醇/乙酸乙酯(150 mL∶30 mL)的混合溶剂中(丙醇∶乙酸乙酯=5∶1,升温50~60℃)。随后,将1-乙基 -3-甲基咪唑溴盐(12 mg/1 mmol/2 mL热蒸馏水)加入,当残留物紫色消失后,趁热过滤,固体用33 mL丙醇/乙酸乙酯混合液(50~60℃)洗涤,合并滤液,加入吗替麦考酚酯纯品0.1克引晶,缓慢冷却至0~5℃,并搅拌1h。过滤晶体,并以45 mL丙醇(0-5℃)洗涤,真空干燥,可得22.7g白色晶体(收率:实际产量/理论产量 =22.7/27.06=84%)。
此工艺具有反应时间短、MPM纯度高、反应收率高、生成成本较低等优点,且解决了终产物颜色问题,具有广阔的工业化应用前景。
[1]Iaccarino L,Rampudda M,Canova M,et al.Mycophenolate mofeti:What is its place in the treatment of autoimmune rheumatic diseases[J].Autoimmunity Reviews,2007,6(3):190-195.
[2]许建军.吗替麦考酚醋在肾移植患者中的应用[J].中国医药导刊,2003,5(3):174-176.
[3]徐敏华.吗替麦考酚醋分散片的研制[J].海峡药学,2007,19(5):11-14.
[4]袁恒杰.吗替麦考酚酸酯临床应用进展[J].天津药学,2012,24(4):62-65.
[5]Nelson P H,Gu C L L,Altison A C,et a1.Morpholnoethyl esters of mycophenolic acid and pharmaceutical compositions[P].US:4753935,1988-06-28.
[6]Knox M,Donegan G,Smith D A,et a1.Direct esterification of mycophenolic acid[P].US:5247083,9,13-09-21.
[7]Laxmi A,Shrikumar S.Microwave synthesis of mycophenolate[P].WO:2004/089946 A1,2004-10-21.
[8]Nitin P,Rakesh M,Anand K,et al.Enzymatic preparation of mycophenolate mofetil[P].WO:2003042393 A1,2003-05-22.
[9]Anindya S,Anand K,Shrikumar S,et al.Methods of producing esters of mycophenolate[P].WO:2000034503,2000-06-15.
[10]Pqride G,Paolo P.A method for the preparation of mycophenolate mofetil by enzimatic transesterification[P].WO:2006024582 A1,2006-03-09.
[11]Miloslav C,Ales H.Method of mycophenolate mofetil preparation[P].WO:02100855 A1,2002-12-19.
[12]Endres F,Zein S,Abedin El.Air and water stable ionic liquids in physical chemistry[J].Phys Chem Chem Phys,2006,8(18),2101-2116.
[13]Bica K,Gaertner P.Applications of chiral ionic liquids[J].Eur J Org Chem,2008,19:235-3250.
[14]Chiappe C,Pieraccini D.Ionic liquids:solvent properties and organic reactivity[J].J Phys Org Chem,2005,18(1):275.
[15]Harper J B,Kobrak M N.Understanding Organic Processes in ionic liquids:achievements so far and challenges remaining[J].Mini-Rev Org Chem,2006,3(3):253-269.
[16]Welton T.Ionic liquids in catalysis[J].Coord Chem Rev,2004,248(3):2459-2477.
[17]Ding J,Armstrong D W.Chiral ionic liquids:Synthesis and applications[J].Chirality,2005,17(5):281-292.
[18]Luo S Z,Zhang L,Cheng J P.Functionalized chiral ionic liquids:a new type of asymmetric organocatalysts and nonclassical chiral ligands[J].Chem Asian J,2009,4(8):1184-1195.
猜你喜欢
分子催化(2022年1期)2022-11-02
环境科学研究(2022年10期)2022-10-19
中南民族大学学报(自然科学版)(2022年3期)2022-05-08
西部交通科技(2022年2期)2022-04-27
食品工业科技(2021年23期)2021-12-16
中国土壤与肥料(2021年5期)2021-12-02
食品安全导刊(2021年21期)2021-08-30
石油沥青(2019年6期)2020-01-16
食品与生活(2017年5期)2017-05-27
东方考古(2016年0期)2016-07-31
