
血清糖化清蛋白定量检测系统的性能验证
2014-09-14 06:16欧阳彬魏玲玲鄢盛恺
中国医药导报 2014年11期
欧阳彬 李 江 魏玲玲 鄢盛恺,▲
1.广东省河源市人民医院检验科,广东河源 517000;2.中日友好医院检验科,北京 100029;3.北京豪迈生物工程有限公司,北京 101318
糖化清蛋白(glycated albumin,GA)由于本身成分的单一,其测定克服了糖化血清蛋白(GSP)检测方法学的缺陷,可以作为一个短期血糖监测(1~2周)的可靠性指标,是糖化血红蛋白(HbA1c)测定的有益补充[1]。近年来,GA在糖尿病患者,尤其是妊娠糖尿病患者的血糖监测中的应用日益广泛。目前国内外市场上与主流全自动生化分析仪配套使用的GA检测试剂并不多见,而众多国内从事体外诊断(IVD)检测试剂生产的厂家,并不生产与之匹配的检测仪器。对此,临床实验室在使用全自动生化分析仪开放通道与开放试剂配套构成的检测系统之前,必须验证检测系统的精密度、准确度、可报告范围、参考区间等检测性能指标,以证明构成的检测系统性能与试剂厂商声明的一致,符合临床实际要求[2],而验证这些性能指标也是目前实验室认可(ISO 15189)过程中的重要技术要求之一。本文参照美国临床和实验室标准协会(CLSI)颁布的标准化文件及其他文献方法[3-4],对北京豪迈生物工程有限公司(以下简称“豪迈”)自主研发生产的血清GA试剂盒[1]与美国贝克曼库尔特有限公司生产的AU2700全自动生化分析仪(以下简称“AU2700生化分析仪”)组成的配套检测系统进行性能验证。
1 材料与方法
1.1 材料
采集2013年10~12月广东省河源市人民医院及中日友好医院门诊、住院患者和正常健康体检者的血清样本,混合成正常水平(GA水平11%~16%)和异常水平(GA水平>16%)的混合新鲜血清各50 mL;收集异常高浓度(GA水平 >65%)血清样本混合成高值新鲜血清,浓度接近或高于线性检测范围(5 mL);采集健康体检者新鲜血清样本,男女各40份,每份1 mL。
1.2 试剂与仪器
豪迈GA检测试剂盒(批号:HX006、HX005、HX004)。 豪迈 GA 校准品(批号:HX006、HX005),贝克曼库尔特AU2700全自动生化分析仪及配套用清洗剂。
1.3 方法
按照试剂说明书中参数设定仪器参数进行实验分析,每天测定2个水平的常规质控品,按实验室室内质控规则判断质控在控后,进行实验分析。
1.3.1 准确度实验
实验一:使用当前校准品校准检测体系,选取与当前校准品批号不同的另一校准品进行检测,与试剂厂商提供的校准品说明书对照,计算相对偏差[5];实验二:回收试验,将不超过总体积10%的校准品溶液加入混合新鲜血清样本中,测定计算得出回收率[6]。
1.3.2 精密度实验
依据CLSI的EP15-A2文件中的实验方案,验证豪迈GA试剂盒与贝克曼库尔特AU2700全自动生化分析仪共同构成的检测系统的精密度,每天分析1个批次样本,每批次2个浓度,每个浓度重复测定3次,连续测定5 d,计算批内精密度[7]。同时测定3个批次试剂,2个浓度,每个浓度重复测定3次,计算批间精密度,进而计算批内和批间变异系数(CV),并与试剂厂家声明的批内CV和批间CV进行比较。
1.3.3 可报告范围[8]
1.3.3.1 线性范围 选择检测结果达到或接近厂家声明的线性范围上限(H)及下限(L)的新鲜血清,制成高值和低值混合样本,低值为1号样本,高值为6号样本。二者以4∶1的比例混匀为2号样本,两者3∶2混匀为3号样本,2∶3混匀为4号样本,1∶4混匀为 5号样本,混合成6份浓度不同的样本,每个浓度的样本重复测定2次。以预期值为±,测定均值为Y,计算回归方程为Y=bX+a。
1.3.3.2 可报告范围 依据试剂厂家声明的稀释液和稀释倍数,收集3份浓度在线性范围内的高值新鲜血清样本,稀释后分别测定,计算其稀释回收率。
1.3.4 参考区间验证
选择体检健康者,年龄18~61岁,男、女新鲜血清样本各40份,按照CLSI颁布的C28-A2文件中的实验方案[9]进行实验,验证厂商声明的参考区间的正确性。
2 结果
2.1 准确度实验
2.1.1 校准验证实验
使用批号为HX006的校准品对当前检测系统进行校准,对批号为HX005的校准品进行检测,对照被测校准品说明书,计算相对偏差,检测值1为20.0%,检测值2为20.2%,平均值为20.1%,低于预期值(20.6),相对偏差为2.4。GA校准品的相对偏差为2.4%。厂家的声明是,相对偏差应在±15%范围内,相对偏差小于厂家声明,符合要求。
2.1.2 回收实验
分别将批号为 HX006 的校准品 20 μL、50 μL 和100 μL加入1000 μL正常混合新鲜血清中,校准品的标示值为20.6%,分别计算回收率。结果显示,3个样本的回收率分别为104.1%、107.9%和103.2%,均在试剂厂商标示的90.0%~110.0%范围内,表明由豪迈GA试剂盒与AU2700生化分析仪组成的检测系统的准确度符合要求。
2.2 精密度实验
每天分析1个批次,2个浓度的样本,每个浓度重复测定3次,连续测定5 d,计算批内精密度。同时使用3个批次试剂,2个浓度,每个浓度重复测定3次,计算批间精密度。低值样本测定均值为13.4%,验证的CV批内=2.42%,CV批间=2.98%,高值样本测定均值为 24.6%,验证的 CV批内=1.67%,CV批间=1.89%,厂家声明的CV批内≤5%,CV批间≤15%,均在厂家声明的允许范围内。见表2。
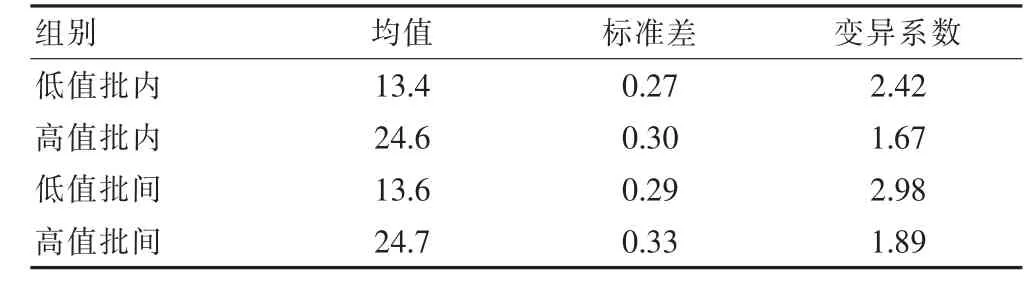
表2 精密度实验结果(%)
2.3 临床可报告范围
2.3.1 线性范围
选取测定值为3.23%的低值样本和测定值为67.50%的高值样本,按不同比例混合,配置成系列浓度血清,并进行双份测定。以验证试剂厂商声明的GA试剂盒的线性范围为3.20%~68.00%。见表3。
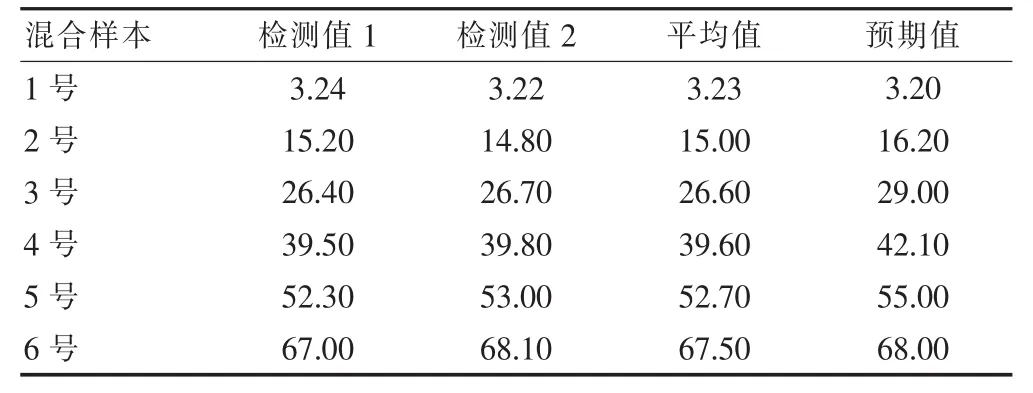
表3 线性范围验证(%)
2.3.2 可报告范围
收集高值血清样本3份,浓度在已经验证的GA测量范围内,用试剂厂商推荐的生理盐水作为稀释液,进行最大2倍稀释,分别重复测定3次,计算3份样本的稀释回收率。结果显示,3份样本的稀释回收率分别为98.12%、96.86%和95.17%,均符合试剂厂商声明的90%~110%的要求范围,故验证的临床可报告范围符合试剂厂商的3.2%~68.0%。
2.4 参考区间
40份健康成年人新鲜血清样本测定结果显示,所有检测值均在试剂厂商提供的参考区间之内(11%~16%),考虑到本实验室的服务人群与厂商提供参考区间的人群相近,检测方法具有可比性,故采用试剂厂商提供的参考区间进行临床应用是符合实际需求的。
3 讨论
根据目前国内外普遍的做法及实验室认可的相关要求,在用于临床检验之前,对于实验室自己构建,或是配套生化检测系统应进行评估并确认相关性能参数,包括准确度、精密度、可报告范围和参考区间,以保证所构建的检测系统符合临床要求,证实其可以给出满意的测定结果。利用简便、有效的方法验证厂家声明的性能参数,并对所用检验系统和程序进行评价,这也是实验室质量管理体系中的重要组成部分[10]。
本实验采用两种方式验证所构建系统的准确度,包括系统间比对试验和系统内有证参考品来验证。本研究中,采用厂家提供定值校准品进行测定及回收实验进行准确度验证,用以验证豪迈GA检测试剂盒与AU2700生化分析仪组合构成的检测系统准确度。结果显示,通过校准后的检测系统对试剂厂商生产的校准品进行测定的相对偏差为2.4%。符合所声明的相对偏差应在±15%范围内。使用3个水平校准品的进行回收试验,回收率分别为104.1%、107.9%和103.2%,均在厂商声明的90%~110%范围内。通过上述两个实验的结果,显示本检测系统准确度符合要求。
通过精密度实验验证得到的批内和批间CV均低于试剂厂商说明书标示的标准,低值样本测定均值为 13.4%,验证的 CV批内=2.01%,CV批间=2.13%,高值样本测定均值为24.6%,验证的CV批内=1.22%,CV批间=1.38%,均在厂家声明的CV批内≤5%、CV批间≤ 15%的允许范围内,说明本检测系统具有良好的精密度。
采用不同稀释比例制成的不同浓度的系列样本进行线性范围验证,发现试剂厂商声明的线性范围(3.20%~68.00%)及最大稀释倍数实用可靠,表3结果显示,线性回归方程为Y=0.986X-1.005,r2=0.998,斜率在0.97~1.03之间,截距接近于0,可认为该检测系统在线性范围内具有良好的线性检测性能,所以可以使用生理盐水稀释超出线性分析范围的样本。按试剂说明书推荐的方法对3份高值样本进行稀释测定并计算回收率,显示3份样本的回收率分别为98.12%、96.86%和95.17%,均符合试剂厂商声明的90%~110%范围,说明试剂说明书所标示的线性范围正确有效。
正确可靠的参考区间是临床实验室开展常规检测工作的必要条件之一,也是临床实验室认可工作的基本要求。通常可以根据专业教科书或厂商说明书等资料制定参考区间,但使用不同检测系统之前应进行参考区间验证,看是否可作为临床实验室实际使用的参考区间。试剂厂商提供的中国人群的GA参考区间为11%~16%,考虑到本实验室的服务人群与豪迈厂家建立参考区间的人群相近,检测方法具有可比性,通过验证实验表明,40例样本的检测值均在试剂厂商提供的参考区间(11%~16%)之内,所以采用此参考区间作为临床实验室实际应用符合要求。
上述实验结果显示,由豪迈GA检测试剂盒和与贝克曼库尔特AU2700生化分析仪构成的配套检测系统的性能指标,包括准确度、精密度、线性范围和临床可报告范围,均与豪迈公司试剂说明书声明的一致,也说明豪迈公司提供的参考区间可以转移使用。上述验证实验表明,本自建检测系统的性能可以满足临床需求,所采用的验证方案简便、可行,可推广用于临床生化定量检测工作中对于各种类型自建或配套检测系统的性能验证。
[1]刘常同,黄萍.糖化清蛋白在糖尿病早期诊断中的应用价值[J].现代检验医学杂志,2012,27(5):121-123.
[2]U.S.Department of Health and human services,centers for medicare&medicaid services.Clinical laboratory improvement amendments of 1988:final rule [S].Fed Register,2003:3704.
[3]黄小兵,刘光明,张伟坚.罗氏全自动生化分析仪自建生化检测系统的性能验证及评价[J].海南医学,2013,24(16):2417-2419.
[4]何国坚,胡劲辉,肖庆.半胱氨酸蛋白酶抑制剂C透射比浊检测试剂盒性能验证[J].检验医学与临床,2013,10(23):3155-3157.
[5]张葵.定量检测系统方法学性能验证实验的基本方法[J].临床检验杂志,2009,27(5):1047-1049.
[6]王治国.临床检验方法确认与性能验证[M].北京:人民卫生出版社,2009:242-297.
[7]Clinical and Laboratory Standards Institute.User verification of performance for precision and trueness:approved guideline second edition[S].EP15-A2(e),CLSI,2005.
[8]杨有业,张秀明.临床检验方法学评价[M].北京:人民卫生出版社,2009:175-195.
[9]CLSI.C28-A2.How to define and determine reference intervals in the clinical laboratory[S].Approved Guideline-Second Edition,2000.
[10]王治国.临床检验方法确认与性能验证[M].北京:人民卫生出版社,2009:242-297.
猜你喜欢
中外玩具制造(2021年2期)2021-02-07
检验医学与临床(2020年1期)2020-01-10
中国医疗器械信息(2018年23期)2019-01-14
汽车观察(2018年10期)2018-11-06
大医生(2017年9期)2017-03-22
系统管理学报(2016年5期)2016-09-03
应用海洋学学报(2015年2期)2015-11-22
现代检验医学杂志(2015年6期)2015-02-06
湖南文理学院学报(自然科学版)(2014年4期)2014-05-13
