
T7噬菌体尾蛋白P11的表达、纯化及鉴定
2014-08-31 09:04赵翠娇宁保安范献军李桂敏高志贤1
郑州大学学报(医学版) 2014年4期
赵翠娇,宁保安,范献军,李桂敏,高志贤1,#
1)郑州大学公共卫生学院营养与食品卫生学教研室 郑州 450001 2)军事医学科学院卫生学环境医学研究所 天津300050
T7噬菌体尾蛋白P11的表达、纯化及鉴定
赵翠娇1),宁保安2),范献军2),李桂敏2),高志贤1,2)#
1)郑州大学公共卫生学院营养与食品卫生学教研室 郑州 450001 2)军事医学科学院卫生学环境医学研究所 天津300050
#通讯作者,男,1966年5月生,博士,研究员,研究方向:食品安全,E-mail:gaozhx@163.com
T7噬菌体;P11蛋白;多克隆抗体
目的:表达、纯化T7噬菌体尾蛋白P11并进行鉴定。方法PCR扩增T7噬菌体尾蛋白P11基因片段,EcoRⅠ和XhoⅠ双酶切后连接表达载体pTIG-TRX,重组表达载体转化大肠杆菌BL21(DE3),经IPTG诱导表达,纯化目的蛋白,SDS-PAGE分析P11蛋白的相对分子质量大小,并多次免疫新西兰大白兔制备其多克隆抗体,Western blot鉴定蛋白活性。结果PCR扩增P11基因后,成功诱导表达了含6×His标签的P11蛋白,SDS-PAGE显示所表达蛋白的相对分子质量约为25 000,通过多次免疫获得了抗P11的多克隆抗体并对其纯化;制备的多克隆抗体能够识别T7噬菌体和P11蛋白,具有较高特异性。结论成功表达了P11蛋白并制备了其多克隆抗体,为T7噬菌体展示系统的应用奠定了基础。
T7噬菌体展示系统具有操作简便、复制周期短、容易培养、生长迅速、系统稳定、不需要分泌到周质或膜外而直接展示的优点,因此,T7噬菌体展示已发展为一种常见的展示系统,被广泛应用于筛选基因工程抗体[1-3]。T7噬菌体衣壳蛋白主要包括头蛋白P10A、P10B,颈圈蛋白P8,尾蛋白P11、P12和尾丝蛋白P17[4-5]。P11蛋白位于T7噬菌体的尾部。将制备的抗P11蛋白多克隆抗体应用于噬菌体ELISA筛查,可使得融合于P10的蛋白或多肽更加有效地展示。为了进一步开展T7噬菌体展示基因工程抗体的研究,作者采用原核表达系统表达P11蛋白并制备出高效价的多克隆抗体。
1 材料与方法
1.1主要试剂与实验动物模板T7 Select 10-3b、表达载体pTIG-TRX、克隆菌株E.coliDH5α、表达菌株BL21(DE3)由)郑州大学公共卫生学院营养与食品卫生学教研室实验室保存。质粒pMD18-T购自大连TaKaRa公司。T4 DNA连接酶、限制性内切酶EcoRⅠ和XhoⅠ购自Fermentas公司。胶回收试剂盒、质粒DNA提取试剂盒购自美国Omega公司。BCA蛋白浓度测定试剂盒购自北京索莱宝科技有限公司。清洁级健康新西兰大白兔由军事医学科学院实验动物中心提供。
1.2重组克隆载体pMD18-T-P11的构建P11蛋白基因片段的特异性上游引物为5’-GCGGAAT TCTAAATGCGCTCATACGATATGAAC-3’,下游引物为5’-GCCCTCGAGGCGAGTCAGTAGACCAG-3’,以T7 Select 10-3b为模板,进行PCR扩增。反应体系共20 μL。扩增条件:94 ℃预变性5 min;94 ℃变性30 s,54 ℃退火30 s,72 ℃延伸1 min,30 个循环;72 ℃延伸7 min;4 ℃保存。15 g/L琼脂糖凝胶电泳鉴定扩增产物并回收。回收后的目的基因片段末端加A后与克隆载体pMD18-T连接,而后将连接产物转入氯化钙法制备的大肠杆菌DH5α感受态,并接种到含有氨苄青霉素(0.1 g/L)的LB固体培养基,37 ℃温箱培养12~16 h,挑取单菌落接种于含氨苄青霉素(0.1 g/L)的LB液体培养基中,37 ℃振荡培养过夜,分别进行菌落PCR和质粒双酶切鉴定阳性克隆子,双阳性克隆子送往北京Invitrogen公司测序。
1.3重组pTIG-TRX-P11表达载体的构建选择测序结果正确的菌株提取质粒,用EcoRⅠ和XhoⅠ双酶切出目的基因,15 g/L琼脂糖凝胶电泳酶切产物并回收,回收后的目的基因与经EcoRⅠ和XhoⅠ双酶切的表达载体pTIG-TRX连接,连接产物转化氯化钙法制备的BL21(DE3)感受态,并接种到含有氨苄青霉素(0.1 g/L)的LB固体培养基,37 ℃温箱培养12~16 h,挑取单菌落接种于含氨苄青霉素(0.1 g/L)的LB液体培养基中,37 ℃振荡培养过夜,分别进行菌落PCR和双酶切鉴定阳性克隆子。
1.4重组P11蛋白的表达及纯化阳性表达菌株过夜活化后以体积比1:100接种于含0.1 g/L氨苄青霉素的LB液体培养基,37 ℃振荡培养3 h至A600 nm约为0.6,加入异丙基-β-D硫代半乳糖苷(IPTG)至终浓度为0.001 mol/L,30 ℃振荡培养5 h后收集菌体。用缓冲液A(0.02 mol/L PBS缓冲液,0.5 mol/L NaCl,pH 7.4)重悬菌体,超声破碎后离心收集上清并用3倍A液稀释,加载到A液预平衡的Ni亲和柱,上样完毕后用A液平衡,用含0.02 mmol/L咪唑的A液洗脱杂蛋白,再分别用含0.05、0.10 mmol/L咪唑的A液洗脱目的蛋白,经SDS-PAGE进行鉴定。纯化得到的P11蛋白经HitrapTMDesalting柱去除咪唑,-20 ℃保存。
1.5动物免疫取健康雌性新西兰大白兔3只,体重2.5 kg左右,免疫前1周于耳缘静脉采血留作阴性血清。第一次基础免疫取0.5 mg纯化后的P11蛋白与等体积的弗氏完全佐剂充分乳化,于家兔背部四点注射,每点注射1 mL,以后每隔2个星期进行1次基础免疫,剂量加倍且用弗氏不完全佐剂乳化,从第3次免疫开始,每次免疫后1周于家兔耳缘静脉采血,用间接ELISA测定血清效价情况。当效价不再上升,进行心脏取血,采血前3 d进行一次不加佐剂的翻倍剂量加强免疫。
1.6多克隆抗体的纯化及鉴定取抗血清用醋酸盐缓冲液(0.06 mol/L,pH 4.0)稀释4倍并调节pH至4.5,缓慢滴加辛酸[6],4 ℃搅拌0.5 h后静置2 h,4 ℃离心20 min(10 000 r/min)收集上清,经0.45 μm滤膜过滤后加入PBS(0.1 mol/L)并调节pH至7.4,于冰水浴中边搅拌边缓慢加入等体积的饱和硫酸铵(pH 7.4),使其成45%的饱和度,继续搅拌0.5 h而后静置2 h,4 ℃离心20 min(10 000 r/min),沉淀溶于PBS,先用双蒸水透析1 h,再用PBS(0.01 mol/L)透析12 h,4 ℃离心10 min,上清即为纯化后所得多抗。抗体纯化效果通过SDS-PAGE进行测定,并根据其结果对加入的辛酸量进行调整,纯化得到的多克隆抗体的浓度通过BCA蛋白定量进行测定。采用Western blot方法鉴定多克隆抗体活性。纯化后的P11蛋白进行120 g/L SDS-PAGE分离,电转移至PVDF膜,50 g/L脱脂奶粉过夜封闭,纯化后多克隆抗体作为一抗(1:10 000稀释),37 ℃孵育1 h,羊抗兔IgG作为二抗(1:2 000稀释),37 ℃孵育1 h,ECL显色试剂进行显色,压胶片显影。
2 结果
2.1P11蛋白基因片段的扩增及重组克隆载体的鉴定经鉴定,pMD18-T-P11构建成功(图1)。
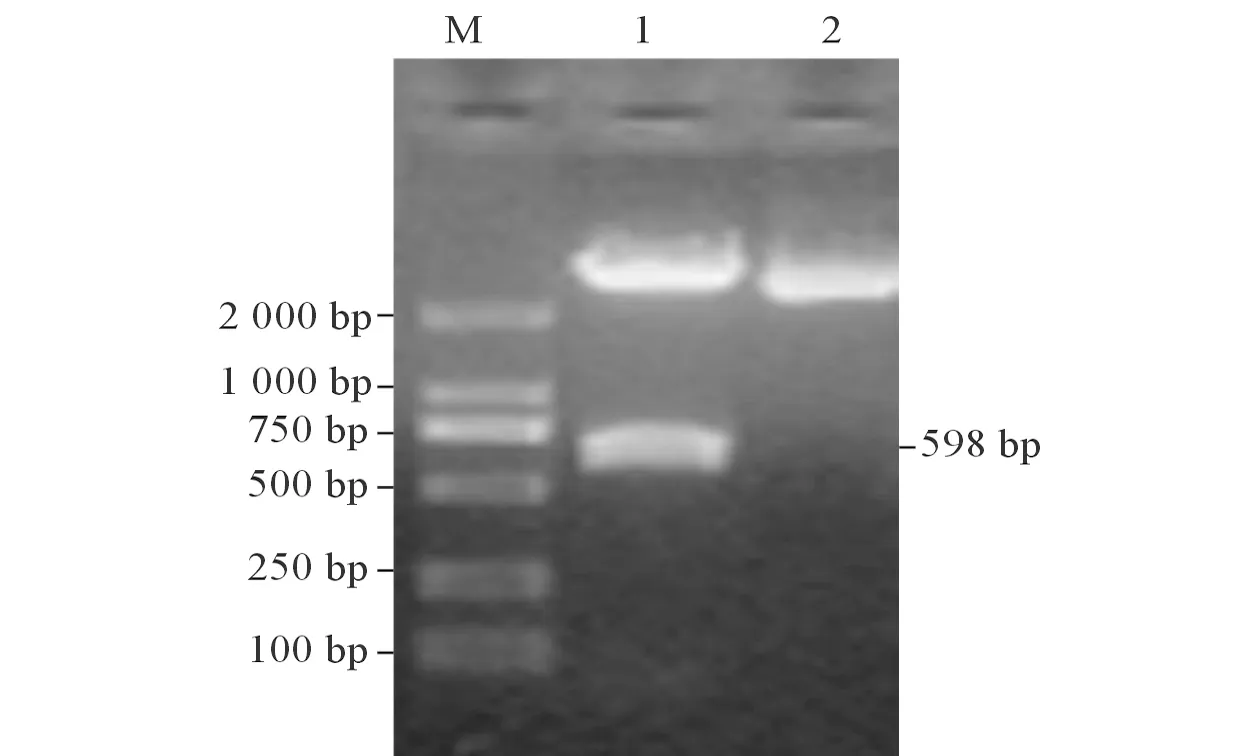
图1 重组克隆载体pMD18-T-P11的酶切鉴定
M:Marker;1:双酶切后的pMD18-T-P11;2:pMD18-T-P11。
2.2重组表达载体的鉴定菌落PCR和双酶切鉴定阳性克隆子结果见图2。
2.3重组P11蛋白的表达、纯化SDS-PAGE电泳结果显示,阳性菌株经IPTG诱、导纯化后,其细菌裂解液在约25 000出现高纯度的蛋白条带,与预期蛋白相对分子质量相符,见图3。
2.4多克隆抗体的纯化及鉴定见图4、5。经条件优化,纯化出的多克隆抗体条带清晰(图4),且经ELISA鉴定其结合活性没有受到影响。Western blot结果显示:在相对分子质量为25 000处T7噬菌体蛋白和6×His-P11蛋白有一条明显的条带,而空质粒即pTIG-TRX菌株诱导蛋白则无此反应(图5)。
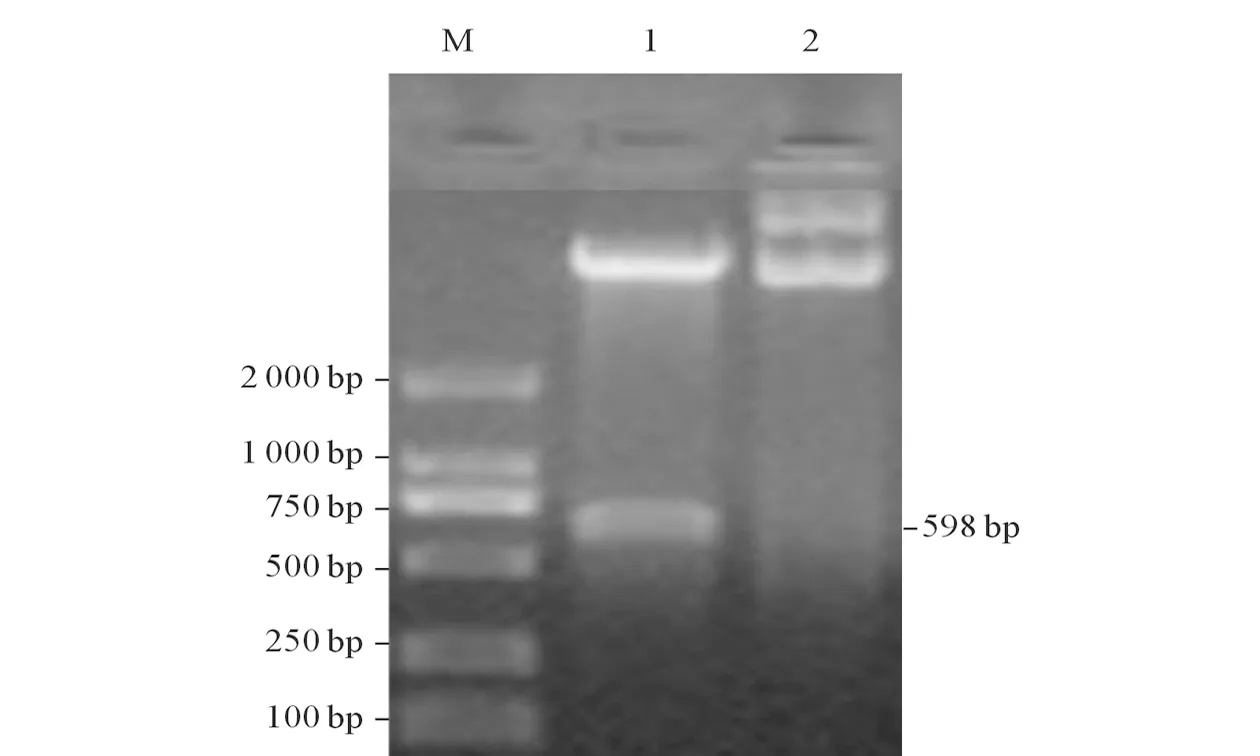
图2 重组克隆载体pTIG-TRX-P11的酶切鉴定
M:Marker;1:双酶切后的PTIG-TRX-P11;2:未酶切的PTIG-TRX-P11。
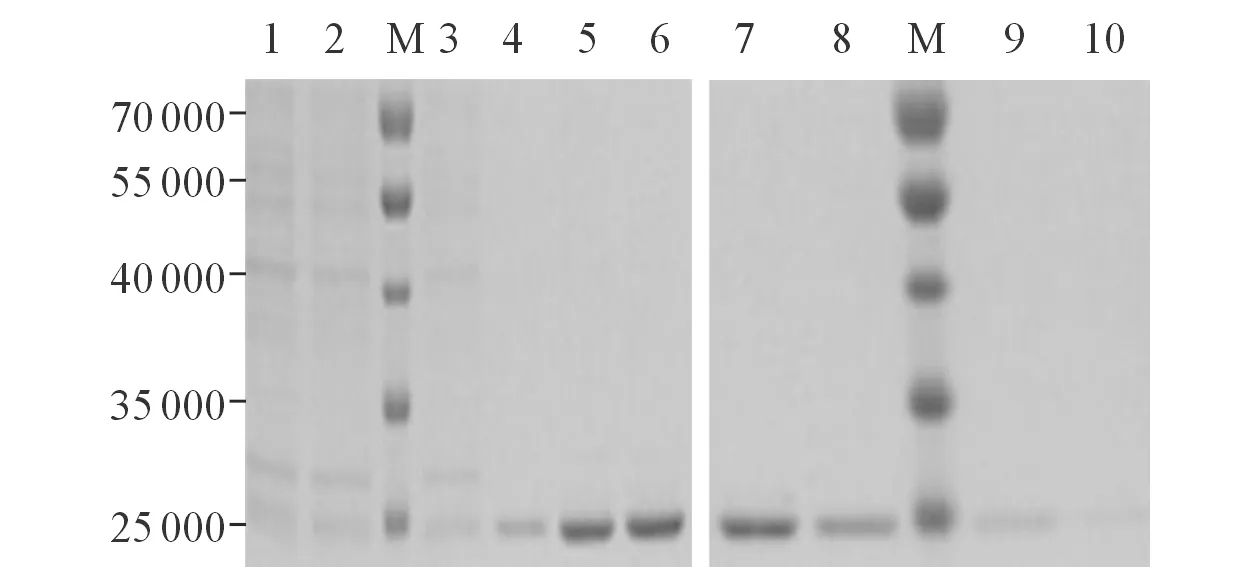
图3 重组P11蛋白的纯化
M:Marker;1~3:含0.02 mol/L咪唑的A液洗脱所得样品;4~6:含0.05 mol/L咪唑的A液洗脱所得样品;7、8:含0.1 mol/L咪唑的A液洗脱所得样品;9、10:含0.5 mol/L咪唑的A液洗脱所得样品。
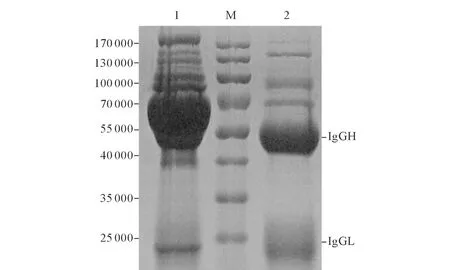
图4 抗P11蛋白多克隆抗体的纯化
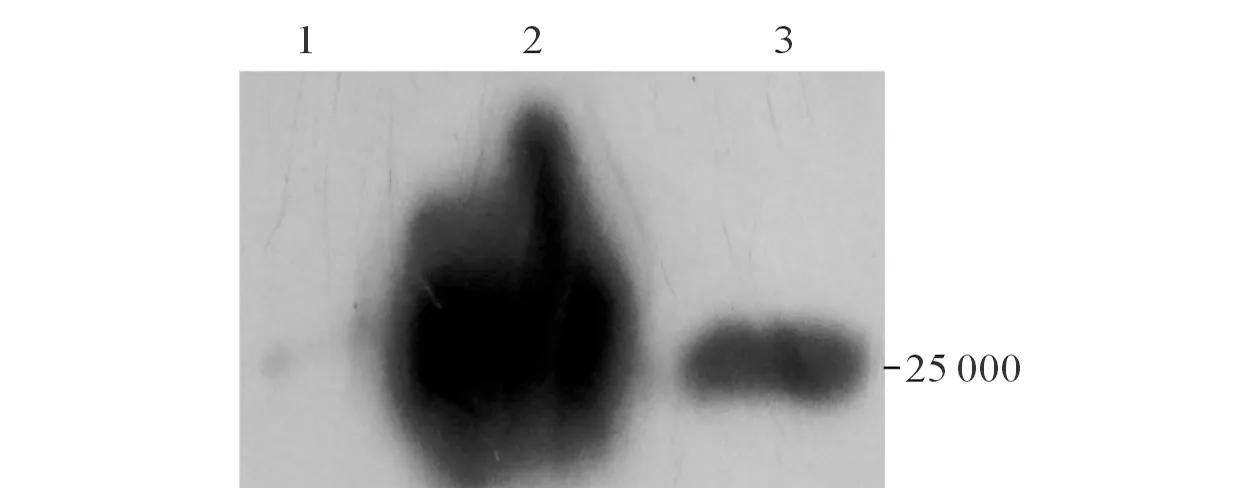
图5 纯化后多克隆抗体的Western blot鉴定
1:含空质粒即pTIG-TRX的菌株诱导表达破碎后上清;2:P11蛋白;3:T7噬菌体。
3 讨论
T7噬菌体展示系统能够较完好地保持所展示蛋白的构象,所筛选出的结合蛋白与其自然状态较为接近[7],与M13噬菌体系统所展示的蛋白相比,具有更好的多样性,因此T7噬菌体展示系统在一定程度上弥补了丝状噬菌体的不足[8],近几年在抗体筛选中应用越来越广泛。
为了开展T7噬菌体展示筛选基因工程抗体的研究,一种高质量纯化的P11蛋白和高效的抗体对于后续的ELISA筛查是必不可少的。为了促进P11蛋白的高效表达,该实验构建了原核表达载体pTIG-TRX,此表达载体是由pET-22b改造而来,即在pET-22b内部插入一段TRX的基因序列,使其能够与目的基因进行共表达。TRX即硫氧还蛋白,它是一种相对分子质量为12 000的氧化还原蛋白,普遍存在于生物体内,其作为原核生物的组成蛋白,虽不属于分子伴侣的范畴,但具有与分子伴侣类似的功能,从而促进包涵体的可溶表达[9]。
该研究构建了6×His标记的T7噬菌体尾蛋白P11原核表达载体PTIG-TRX-P11,鉴定其序列正确后转入大肠杆菌BL21(DE3)并进行诱导表达。利用6×His标签和镍离子特异性螯合的特性纯化,得到高纯度的目的蛋白。以纯化的P11蛋白为免疫原免疫新西兰大白兔成功获得了抗P11蛋白的多克隆抗体,为后续的T7噬菌体展示筛选基因工程抗体奠定了基础。
[1]Smith GP,Petrenko VA.Phage Display[J].Chem Rev,1997,97(2):391
[2]陈卉爽,范南辉,宁保安.T7噬菌体衣壳蛋白10A的表达、纯化及多克隆抗体的制备[J].解放军医学杂志,2013,12(31):484
[3] Yu X,Zhao P,Zhang W,et al.Screening of phage displayed human liver cDNA library against dexamethasone[J].J Pharm Biomed Anal,2007,45(5):701
[4]Cerritelli ME,Studier FW.Assembly of T7 capsids from independently expressed and purified head protein and scafolding protein [J].J Mol Biol,1996,258(2):286
[5]Son M,Serwer P.Role of exonuclease in the specificity of bacteriophage T7 DNA packaging[J].Virology,1992,190(2):824
[6]陈丹,孙广瑞,柳增善. 辛酸-硫酸铵联合沉淀法在单克隆抗体纯化中的应用[J].安徽农业科学,2007,35(26):8105
[7]Sergeeva A,Kolonin MG,Molldrem JJ.Display technologies:application for the discovery of drug and gene delivery agents[J].Adv Drug Deliv Rev,2006,58(15):1622
[8]Krumpe LR,Atkinson AJ,Smythers GW.T7 lytic phage-displayed peptide libraries exhibit less sequence bias than M13 filamentous phage-displayed peptide libraries Proteomics[J].Proteomics,2006,6(15):4210
[9]LaVallie ER,DiBlasio EA,Kovacic S, et al.A thioredoxin gen fusion expression system that circumvents inclusion body formation inE.colicytoplasm[J].Biotechnology,1993,11(2):187
(2013-10-29收稿 责任编辑李沛寰)
Expression,purification and identification of T7 bacteriophage tail protein P11
ZHAOCuijiao1),NINGBao'an2),FANXianjun2),LIGuimin2),GAOZhixian1,2)1)DepartmentofNutritionandFoodHygiene,CollegeofPublicHealth,ZhengzhouUniversity,Zhengzhou450001
2)InstituteofHealth&EnvironmentMedicine,AcademyofMilitaryMedicalScience,Tianjin300050
T7 bacteriophage;P11 protein;polycolonal antibody
Aim: To express,purify and identify capsid tail protein P11 of T7 bacteriophage. Methods: Gene of protein P11 was amplified by PCR , digested with restriction endonucleaseEcoRⅠandXhoⅠand then inserted into the expression vector pTIG-TRX.The recombinant vector was transformed toE.coliBL21(DE3).After IPTG induction,the recombinant protein was purified by metal chelate chromatography.Its molecular weight was identified by SDS-PAGE and its activity was analysed by Western blot.The recombinant protein P11 was used to immune rabbit. Finally,the rabbit polycolonal antibody against protein P11 were obtained. Results: Gene of protein P11 was amplified by PCR.The recombinant protein P11 with 6×His tag has been successfully expressed after induction. SDS-PAGE analysis suggested that the molecular weight of the expressed protein was approximately 25 000. Western blot analysis suggested that the polycolonal antibodidy could recognize T7 phage and P11.Conclusion: We have purified P11 fusion protein and prepared the rabbit polycolonal antibody against protein P11 successfully,which lays foundation for the application of T7 phage display.
10.13705/j.issn.1671-6825.2014.04.017
R392.11
猜你喜欢
——一道江苏高考题的奥秘解读和拓展
中学生物学(2022年7期)2022-09-07
成都医学院学报(2022年4期)2022-08-19
昆明医科大学学报(2022年2期)2022-03-29
植物保护(2021年4期)2021-11-12
江西农业学报(2021年4期)2021-04-20
中华养生保健(2020年3期)2020-11-16
三农资讯半月报(2020年11期)2020-06-21
科学24小时(2020年4期)2020-05-14
农药科学与管理(2019年9期)2019-11-23
