
微波辐射合成2-乙氧羰基甲硫基-4-取代苯基亚氨基-4H-1-萘酮类化合物*
2014-08-30 02:07:36汤雁波肖志艳
合成化学 2014年5期
武 波,汤雁波,肖志艳
(中国医学科学院 北京协和医学院 药物研究所 活性物质发现与适药化研究北京市重点实验室,北京 100050)
·快递论文·
微波辐射合成2-乙氧羰基甲硫基-4-取代苯基亚氨基-4H-1-萘酮类化合物*
武 波,汤雁波,肖志艳
(中国医学科学院 北京协和医学院 药物研究所 活性物质发现与适药化研究北京市重点实验室,北京 100050)
以对取代苯胺与2-乙氧羰基甲硫基-1,4-萘醌为底物,采用微波合成法合成了3个新型的蛋白酶体抑制剂的关键中间体——2-乙氧羰基甲硫基-4-苯基亚氨基-4H-1-萘酮类化合物(3a~3c),其结构经1H NMR和1D NOE表征。在最佳反应条件[THF为溶剂,TiCl4为催化剂,于60℃微波(240W)反应15min]下,2-乙氧羰基甲硫基-4-(4-硝基苯基)亚氨基-4H-1-萘酮(3a)的收率84.2%。
萘醌;酮胺缩合;萘酮;蛋白酶体抑制剂;微波合成
蛋白酶体是目前抗肿瘤药物研究的重要靶点之一,蛋白酶体抑制剂Bortezomib和Carfilzomib先后作为多发性骨髓瘤的治疗药物被FDA批准上市后,该领域的研究更为活跃[1-2]。Lawrence等[3]报道了萘醌类蛋白酶体抑制剂PI-083(Chart 1),以及由萘醌衍生得到的羟基萘类蛋白酶体抑制剂PI-8182(Chart 1)[4-6]的合成。
本课题组[7]曾以PI-083为模板分子,对其2-位进行结构多样性的衍生化,当2-位引入4-取代苯胺时,抗肿瘤活性较好,其中引入对硝基苯胺所得化合物对蛋白酶体的抑制活性优于PI-083。基于此思路,本文以PI-8182为模板分子,在其4-位引入4-取代苯胺(Ⅰ),以期获得结构新颖、活性较好的蛋白酶体抑制剂。

对Ⅰ进行逆合成分析(Scheme 1)可得中间体——2-乙氧羰基甲硫基-1,4-萘醌(1)。由萘醌合成1以及由2-乙氧羰基甲硫基-4-苯基亚氨基-4H-1-萘酮类化合物(3)合成目标分子Ⅰ均可参照文献[2-4]方法进行,而由1合成亚胺中间体3则是能否顺利合成Ⅰ的关键。虽然3可由芳胺和萘醌类化合物经酮胺缩合反应而得,但芳胺的活性对反应进行的难易及收率有较大影响,而且使酮胺缩合反应区域选择性地发生在萘醌的4-位羰基也需要适当的反应条件。Matsuo等[8]在研究1,4-萘醌与芳基亚胺二镁试剂反应时发现,除4-位羰基发生酮胺缩合反应形成预期的亚胺类产物外,还有其它产物生成。由此提示,由1合成3反应条件的选择至关重要。
在PI-8182类抑制剂的制备中,Lawrence[4-6]采用微波合成法,由1和磺胺制得磺酰亚胺中间体。该方法虽然具有快速、高效、副反应少等优点,但存在仪器较昂贵、反应规模有限等局限。
为了高效合成3,并扩大反应条件的适用范围,本文参照文献[4-6]报道的微波合成方法,以对取代苯胺(2a~2c)为底物与1反应,合成了3个新型的2-乙氧羰基甲硫基-4-取代-苯基亚氨基-4H-1-萘酮类化合物(3a~3c,Scheme 2),其结构和立体构型经1H NMR和1D NOE表征。并对反应条件进行优化。
1 实验部分
1.1 仪器与试剂
Varian Mercury-300型核磁共振仪(CDCl3为溶剂,TMS为内标);Agilent LC/MSD TOF型液-质联用仪;SHIMADZU LC-20A型高效液相色谱仪;Biotage InitiatorTM Eight EXP型微波合成仪。
1参照文献[4-6]方法合成;其余所用试剂均为分析纯,其中DCM和THF经氢化钙回流重蒸,DMF经分子筛干燥。
1.23a~3c的合成(以3a为例)
在20mL微波反应管中加入THF 5mL,155.2mg(0.2mmol)和对硝基苯胺(2a)27.6mg(0.2mmol),搅拌使其溶解;于0℃加入TiCl437.9mg(0.2mmol)的THF(0.2mL)溶液和三乙胺0.06mL,加毕,微波(240W)反应15min。用DCM 50mL稀释,加水30mL淬灭反应,分液,水相用DCM(3×20mL)萃取,合并有机相,用无水硫酸钠干燥,减压浓缩后经硅胶柱层析[洗脱剂:A=V(石油醚)∶V(乙酸乙酯)=30∶1~10∶1]纯化,混合溶剂(A=50∶1)重结晶得3a55mg。
用类似方法合成3b~3c。
3a:橙色针状晶体,收率69%,m.p.157℃~159℃;1H NMRδ: 8.42(d,J=7.5Hz,1H,ArH),8.32(d,J=8.4Hz,2H,ArH),8.20(d,J=7.5Hz,1H,ArH),7.80~7.68(m,2H,ArH),7.03(d,J=8.4Hz,2H,ArH),6.77(s,1H,a-H),4.08(q,J=7.2Hz,2H,CH2CH3),3.42(s,2H,b-H),1.18(t,J=6.9Hz,3H,CH3);ESI-MSm/z(%): 397.1{[M+H]+}。
3b:暗红色针状晶体,m.p.129℃~131℃;1H NMRδ: 8.56(d,J=7.5Hz,1H,ArH),8.19(d,J=7.5Hz,1H,ArH),7.70(m,2H,ArH),7.28(d,J=8.4Hz,2H,ArH),7.08(s,1H,a-H),6.93(d,J=8.4Hz,2H,ArH),4.09(q,J=7.2Hz,2H,CH2CH3),3.49(s,2H,b-H),2.55(m,1H,-H),1.96~1.78(m,5H,a′-H),1.57~1.21(m,5H,a′-H),1.16(t,J=7.2Hz,3H,CH3);ESI-MSm/z(%): 434.3{[M+H]+}。
3c:红棕色针状晶体,m.p.163℃~165℃;1H NMRδ: 8.52(d,J=7.5Hz,1H,ArH),8.19(d,J=7.5Hz,1H,ArH),7.69(m,2H,ArH),7.11(s,1H,a-H),6.98(m,4H,ArH),4.11(q,J=7.2Hz,2H,CH2CH3),3.87(s,3H,OCH3),3.50(s,2H,b-H),1.19(t,J=7.2Hz,3H,CH2CH3);ESI-MSm/z(%): 382.1{[M+H]+}。
2 结果与讨论
2.1 表征
Lawrence[5]通过比较萘醌1与由其经与磺胺反应获得的磺酰亚胺的3-H的吸收峰位置变化,推断4-位羰基形成了磺酰亚胺。然而,1H NMR分析表明,1与3a的a-H吸收峰位置差异很小(6.71vs6.77)。为了更明确地确定3的结构及其顺反异构问题,我们对3进行了1D NOE分析。结果表明,萘醌环上的a-H与取代苯胺上的1′-H存在NOE(Chart 2),由此判断,与取代苯胺发生酮胺缩合反应的是4-位羰基,而所形成的碳-氮双键为E-型。
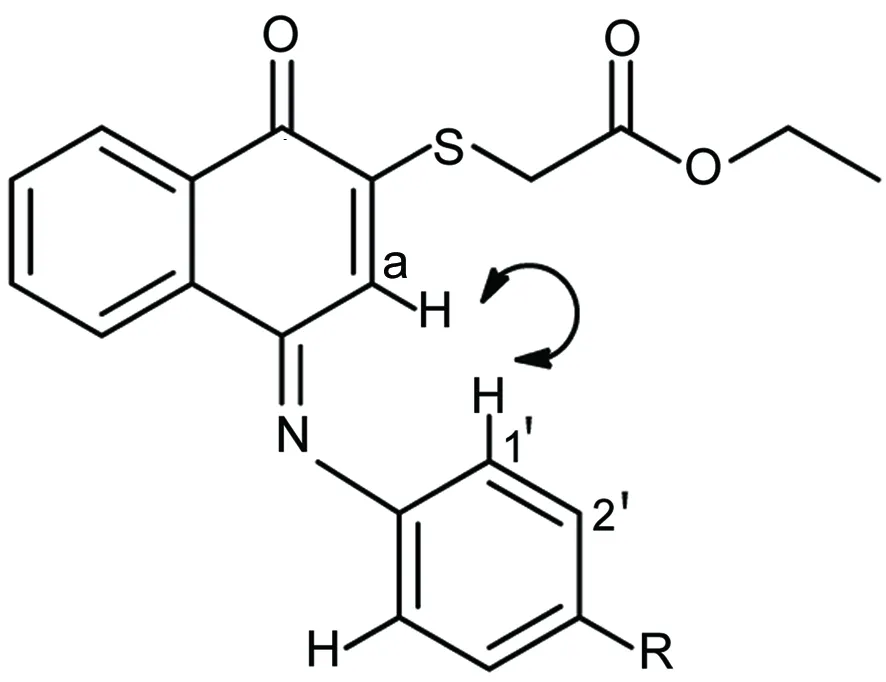
Chart 2
2.2 反应条件优化
以合成3a为模板反应,考察微波合成法中反应时间、溶剂及催化剂对反应的影响,寻找最佳反应条件,结果见表1。由表1可见,在10.2mmol,三乙胺0.4mmol,催化剂TiCl40.2mmol,DCM 5mL反应条件下,最优反应时间为15min(No.2),略短于文献[4-6]方法中的最优反应时间(20min),可能由于2a的亲核能力较磺胺更强;THF为溶剂时对提高反应收率有利,收率84.2%(No.6);另外,TiCl4的催化作用优于AlCl3。
微波合成法的最优反应条件为:THF为溶剂,TiCl4为催化剂,于60℃微波(240W)反应15min(No.9)。
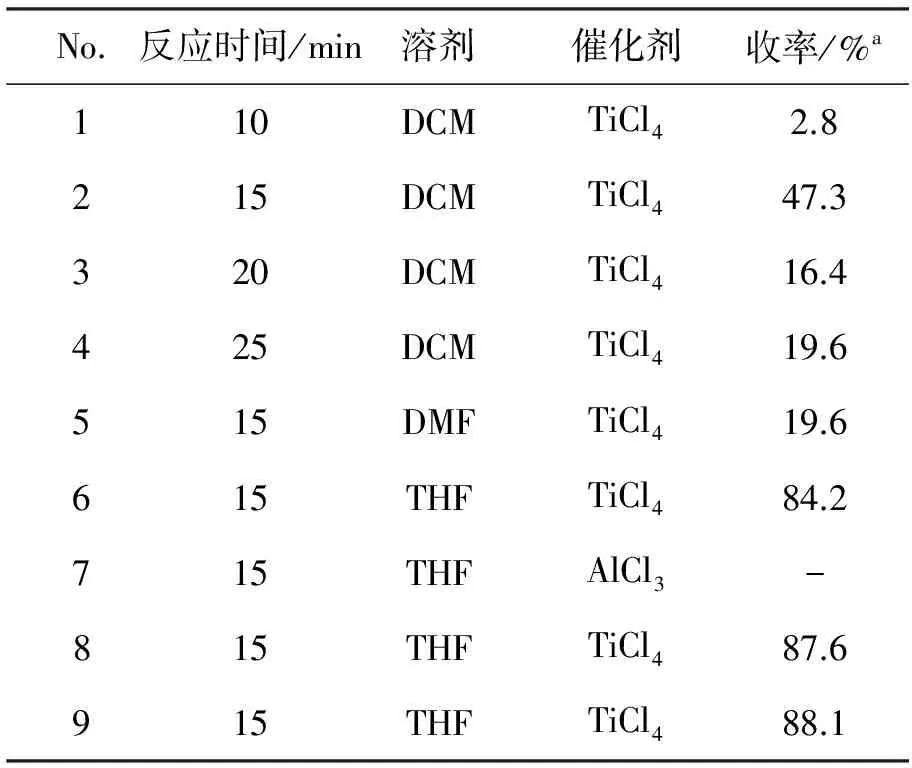
表1 微波合成法的反应条件优化*Table1 Optimization of microwave reaction conditions
*10.2mmol,三乙胺0.4mmol,催化剂0.2mmol,溶剂5mL,其余反应条件同1.2;aHPLC外标法测定
参照微波合成法,其余反应条件同1.2,采用室温搅拌法由1合成3a,考察了反应时间对收率的影响,实验结果表明:在室温搅拌反应中,反应时间分别为40min和10h时,收率分别为69%和74%,差异不大,说明40min时反应已基本进行完全,可以较好的收率获得目标分子。
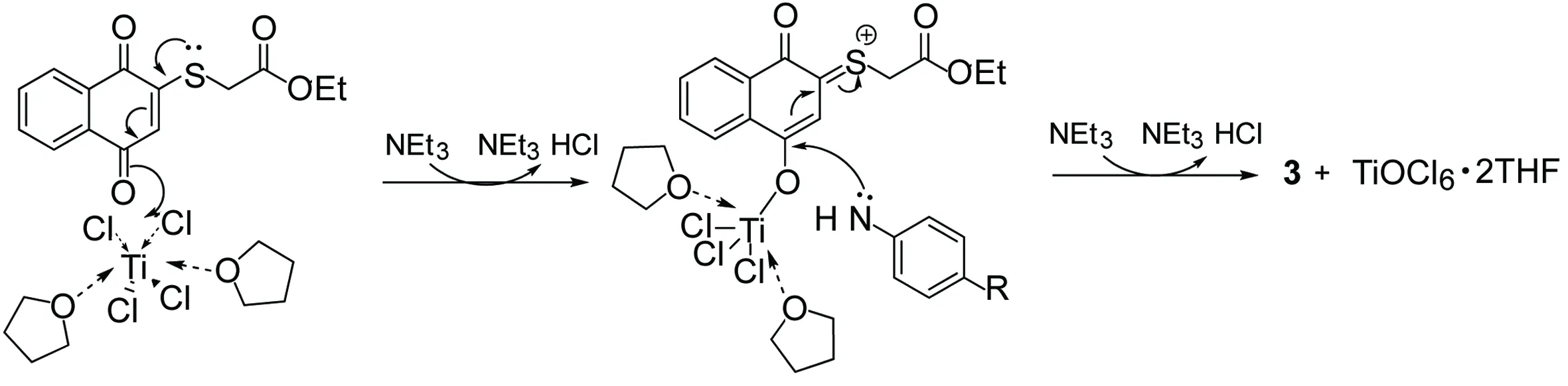
Scheme 3
2.3 催化机理
TiCl4-Et3N体系广泛应用于磺酰亚胺类化合物的合成,TiCl4-羰基可形成配合物,从而活化酮羰基。文献[5]指出,2-位硫原子上的孤对电子与萘醌体系形成的p-π共轭有利于4-位羰基与TiCl4的配位,从而实现反应的区域选择性。据此可推测TiCl4催化1合成3的催化机理(Scheme 3),可解释本实验中酮胺缩合反应的区域选择性。此外,溶剂THF中氧原子的孤对电子可与TiCl4钛原子的空轨道形成配位键,进一步稳定TiCl4-羰基配合物,从而提高TiCl4作为路易斯酸的催化效率。
3 结论
采用微波合成法合成了3个新型的蛋白酶体抑制剂的关键中间体——2-乙氧羰基甲硫基-4-取代-苯基亚氨基-4H-1-萘酮类化合物,并对反应条件进行了筛选和优化。为具有结构通式Ⅰ的新型蛋白酶体抑制剂的构效关系研究提供了参考。
[1] Kisselev A F,van der Linden W A,Overkleeft H S.Proteasome inhibitors:An expanding army attacking a unique target[J].Chem Biol,2012,19(1):99-115.
[2] Genin E,Reboud-Ravaux M,Vidal J.Proteasome inhibitors:Recent advances and new perspectives in medicinal chemistry[J].Curr Top Med Chem,2010,10(3):232-56.
[3] Kazi A,Lawrence H,Guida W C,etal.Discovery of a novel proteasome inhibitor selective for cancer cells over non-transformed cells[J].Cell Cycle,2009,8(12):1940-1951.
[4] Ge Y,Kazi A,Marsilio F,etal.Discovery and synthesis of hydronaphthoquinones as novel proteasome inhibitors[J].J Med Chem,2012,55(5):1978-98.
[5] Lawrence H,Ge Y,Sebti S M,etal.Proteasome inhibitors for selectivity inducing apoptosis in cancer cells[P].WO 2010005534A2,2010.
[6] Lawrence H,Ge Y,Sebti S M,etal.Proteasome inhibitors having chymotrypsin-like activity[P].WO 2010102286A2,2010.
[7] Xu K,Xiao Z,Tang Y B,etal.Design and synthesis of naphthoquinone derivatives as antiproliferative agents and 20S proteasome inhibitors[J].Bioorg Med Chem Lett,2012,22(8):2772-2274.
[8] Matsuo K,Shiraki R,Okubo M.Reactions of iminodimagnesium reagents with 1,4-quinones:Their structural factors governing the modes of reactions[J].J Phys Org Chem,1994,7:567-577.
Synthesisof2-(Ethoxycarbonylmethylthio)-4-substitued-(pheny-limino)naphthalen-1(4H)-onesunderMicrowaveIrradiation
WU Bo, TANG Yan-bo, XIAO Zhi-yan
(Beijing Key Laboratory of Active Substance Discovery and Druggability Evaluation,Institute of Materia Medica,Chinese Academy of Medical Sciences,Peking Union Medical College,Beijing 100050,China)
Three novel key intermediates for preparation of proteasome inhibitors,2-(ethoxycarbonylmethylthio)-4-substitued-(phenylimino)naphthalen-1(4H)-ones(3a~3c),were synthesized by the reaction ofp-substituted aniline with 2-(ethoxycarbonylmethylthio)-1,4-naphthoquinone under microwave irradiation.The structures were characterized by1H NMR and 1D NOE spectra.The yield of 2-(ethoxycarbonylmethylthio)-4-(4-nitrophenylimino)naphthalen-1(4H)-one(3a)was 84.2% under the optimal reaction conditions[THF as the sovlent,TiCl4as the catalyst,microwave radiation(240W)at 60℃ for 15min].
naphthoquinone;ketoamine condensation;proteasome inhibitor;microwave synthesis
2014-02-27
国家自然科学基金资助项目(81302703)
武波(1989- ),男,汉族,山东泰安人,硕士研究生,主要从事药物化学的研究。
肖志艳,研究员,硕士生导师,E-mail: xiaoz@imm.ac.cn
O625.15;O621.3
A
1005-1511(2014)05-0630-04
猜你喜欢
食品工业(2022年11期)2022-11-28 03:26:48
食品与发酵工业(2022年5期)2022-03-30 09:01:08
中国酿造(2022年1期)2022-02-07 13:09:30
广州化学(2020年6期)2020-12-28 06:53:12
黑龙江八一农垦大学学报(2020年2期)2020-05-06 03:34:48
华东师范大学学报(自然科学版)(2018年2期)2018-05-14 10:27:17
科学中国人(2017年36期)2017-06-09 07:58:26
当代化工研究(2016年9期)2016-03-20 16:22:14
中国病理生理杂志(2015年8期)2015-12-21 12:38:08
四川师范大学学报(自然科学版)(2015年1期)2015-02-28 14:07:30
