
芳香偶氮化合物的合成研究进展*
2014-08-30 02:49张鲁博赵秀秀庞思平李玉川孙成辉李生华
合成化学 2014年5期
张鲁博,赵秀秀,庞思平,李玉川,孙成辉,李生华
(北京理工大学 材料学院,北京 100081)
·综合评述·
芳香偶氮化合物的合成研究进展*
张鲁博,赵秀秀,庞思平,李玉川,孙成辉,李生华
(北京理工大学 材料学院,北京 100081)
综述了芳香偶氮化合物的主要合成方法,包括重氮偶联反应、米尔斯反应、胺的氧化、芳肼的的氧化、硝基化合物的还原偶联反应、重氮盐的二聚反应、叠氮化合物的热解等,并对各方法的优缺点进行了简要评述。参考文献96篇。
芳香偶氮化合物;合成方法;偶联;综述
芳香偶氮化合物具有氮氮双键及庞大的π电子体系,存在顺式和反式两种异构体,在一定光照条件下,顺反式之间可以互相转换,利用这个性质可以制作很多光学材料,应用于光通讯、光调制、传输器件等领域。同时偶氮基是一个发色团,因此芳香偶氮化合物一般都具有颜色,偶氮染料是品种最多、应用最广的一类合成染料[1-2]。近年来众多研究结果表明,一些偶氮液晶膜具有热释电和压电效应,可用于制备各种滤色器、光电设备、光敏感元件、光电池、电子发光设备中的释电薄膜等[3]。此外,引入偶氮键不仅能提高氮含量,生成焓和密度,还能降低化合物的感度,所以芳香偶氮化合物也广泛应用于含能材料领域[4-6]。
对于芳香偶氮化合物的合成,传统的方法有重氮偶联反应和米尔斯反应。近年来,研究人员们又发展出一些新的合成方法,例如芳香胺的氧化反应、芳肼的氧化反应、芳香硝基衍生物的还原偶联反应、重氮盐的二聚反应及叠氮化物的热解反应等等。本文对这些合成方法进行系统地介绍,并对各方法的优缺点进行了简要评述。
1 芳香偶氮化合物的合成方法
1.1 重氮偶联反应
重氮偶联反应是合成芳香偶氮化合物的主要方法,产物多为不对称化合物[7-12]。该反应是芳香伯胺的重氮盐与偶合组分——电荷密度较高的芳香族化合物之间进行的反应。
重氮偶联反应为亲电取代反应,化合物的结构对反应进行难易有很大影响。就重氮盐来讲,芳环上有吸电子取代基,可以加强重氮盐的亲电性,使偶联反应容易进行;而芳环上有给电子取代基时,则由于这些取代基的排斥电子作用,使得氮原子的正电性被部分补偿而降低,重氮盐的稳定性提高,减弱了它的亲电性,偶联反应困难。而对于偶合组分,芳环上有吸电子取代基时可降低其电子云密度,使得重氮离子不易进攻,一般不发生偶联反应;相反如有给电子取代基,特别是强给电子基,例如羟基,氨基等,将使芳环的电子云密度增加,偶联反应容易进行[13-15]。1998年,Haghbeen K等[7]将重氮盐(1)和邻甲氧基苯酚(2)在K2CO3溶液中反应制得芳香偶氮苯(3,Scheme 1),由于重氮盐上硝基的吸电子效应,以及偶合组分2上羟基和甲氧基的给电子效应,该反应的产率很高,达92%。
发生重氮偶联反应时,重氮正离子通常进攻偶合组分供电子基团的对位,对位占据则进攻邻位,邻对位都占据则不再发生反应。2002年,Merrington J 等[8]进行了如下反应,将重氮盐(4)与固定在聚苯乙烯树脂上的磺酸盐进行阴离子交换反应,生成重氮盐(5),然后与对羟基萘磺酸(6)发生反应,由于供电子基团OH的对位被SO3H占据,所以反应发生在邻位,生成偶氮化合物(7,Scheme 2)。
对于重氮偶联反应,介质的pH是很关键的,偶联反应常在弱酸、中性或碱性环境中进行。对于酚类来说,在碱性溶液中易离解为酚氧负氧离子ArO-,它比酚更容易进行亲电取代,但若碱性太强(pH≥10),重氮盐会转化为不活泼的重氮酸盐等而不发生偶联(Scheme 3)[9]。
而芳胺类是在中性或弱酸性介质中进行反应,在此条件下,芳胺以游离胺形式存在,芳环电子云密度增加,有利于偶联反应进行。如果溶液酸性过强,胺变成了铵盐,芳环电子云密度降低,就不再发生偶联反应[9]。
2006年,Shiao Y J等[10]将苯酚(9)和重氮盐(8)在弱碱性的反应介质(EtONa溶液)中反应,该反应极易发生,生成偶氮化合物(10,Scheme 4),产率很高。
重氮盐阴离子的性质也会影响该类反应的发生,如以Cl-作为负离子的重氮盐不稳定,在5℃以上会剧烈分解。而四氟硼酸重氮盐、二磺酰亚胺重氮盐及六氟磷酸重氮盐比较稳定,可以长时间储存,很容易和有机芳香金属试剂发生反应。如芳香四氟硼酸重氮盐(11)和二苯锌反应,生成偶氮化合物(12,Scheme 5),产率72%~95%[11]。
此外,结构复杂、分子量较大的化合物,如卟啉[12]、间环芳烃[16]、杯芳烃[17]也可以进行重氮偶合反应。根据前述理论,杯芳烃作为偶合组分,含有大量强给电子基团——羟基,亲核性增强,有利于发生重氮偶合反应。重氮盐(14)和硫杂杯芳烃(13)在吡啶和四氢呋喃中反应,生成偶氮化合物(15,Scheme 6),产率71%[18]。
采用相似的方法,Parola课题组和Lhotak课题组报道了偶氮化合物(16~20,Scheme 7)的合成[18-19]。


1.2 米尔斯反应
亚硝基芳香化合物和一级芳香胺在冰醋酸的催化下生成芳香偶氮化合物的反应称为米尔斯反应。
其中亚硝基芳香化合物可以由胺或羟胺氧化而得,氧化剂有FeCl3[20],过一硫酸(H2SO5)[21],醋酸/双氧水[22],间氯过氧苯甲酸[23],偶氮二甲酸二乙酯[24]及过氧甲酸[25]等。但胺和羟胺的氧化会带来一些副反应,芳香胺(21)氧化不足则生成芳香羟胺(22),它可与亚硝基化合物(23)反应生成氧化偶氮苯(25);氧化过度则生成硝基化合物(24,Scheme 8),不能进一步发生反应[15]。在很大程度上,当芳香胺的间位或对位有吸电子基团时,胺的反应活性会降低,这时副反应容易发生,且延长反应时间会助长副反应的发生。
如氧化剂Oxone(即2KHSO5·KHSO4·K2SO4)在两相体系H2O/CH2Cl2中可有效氧化芳香胺(26)生成亚硝基化合物(27),然后和芳香胺(28)反应,生成一系列的偶氮衍生物(29,Scheme 9)。该反应的产率很高,因为两相体系可以使水溶性较差的亚硝基化合物和氧化过程中的中间体芳基羟胺分离开来,从而有效避免副反应的发生,提高产率[26]。
但是两相反应体系也有缺点,由于反应过程缓慢,有些胺或者羟胺不稳定,反应过程中会发生分解致使产率下降。1999年,Davey M H等[27]采用t-BuOCl作氧化剂进行均相反应,将羟胺(30)氧化为亚硝基化合物(31),这个氧化反应进行很快,在-78℃就可以发生,要经过适度稀释来防止过度氧化,由此制得的31和对甲氧基苯胺(32)反应生成一系列偶氮化合物(33,Scheme 10)。
该作者还对米尔斯反应的实验机理做了详细解释,认为芳香胺N原子上的孤对电子进攻亚硝基,通过电子转移作用生成化合物(36),化合物36与37之间进行快速转化,在酸性条件下,37脱去一个水分子生成偶氮化合物(38,Scheme 11)[25]。
相对于重氮偶合反应,米尔斯反应对底物的限制较少,无论胺还是亚硝基化合物,无论邻位、间位、对位都可以存在吸电子或者供电子基团。
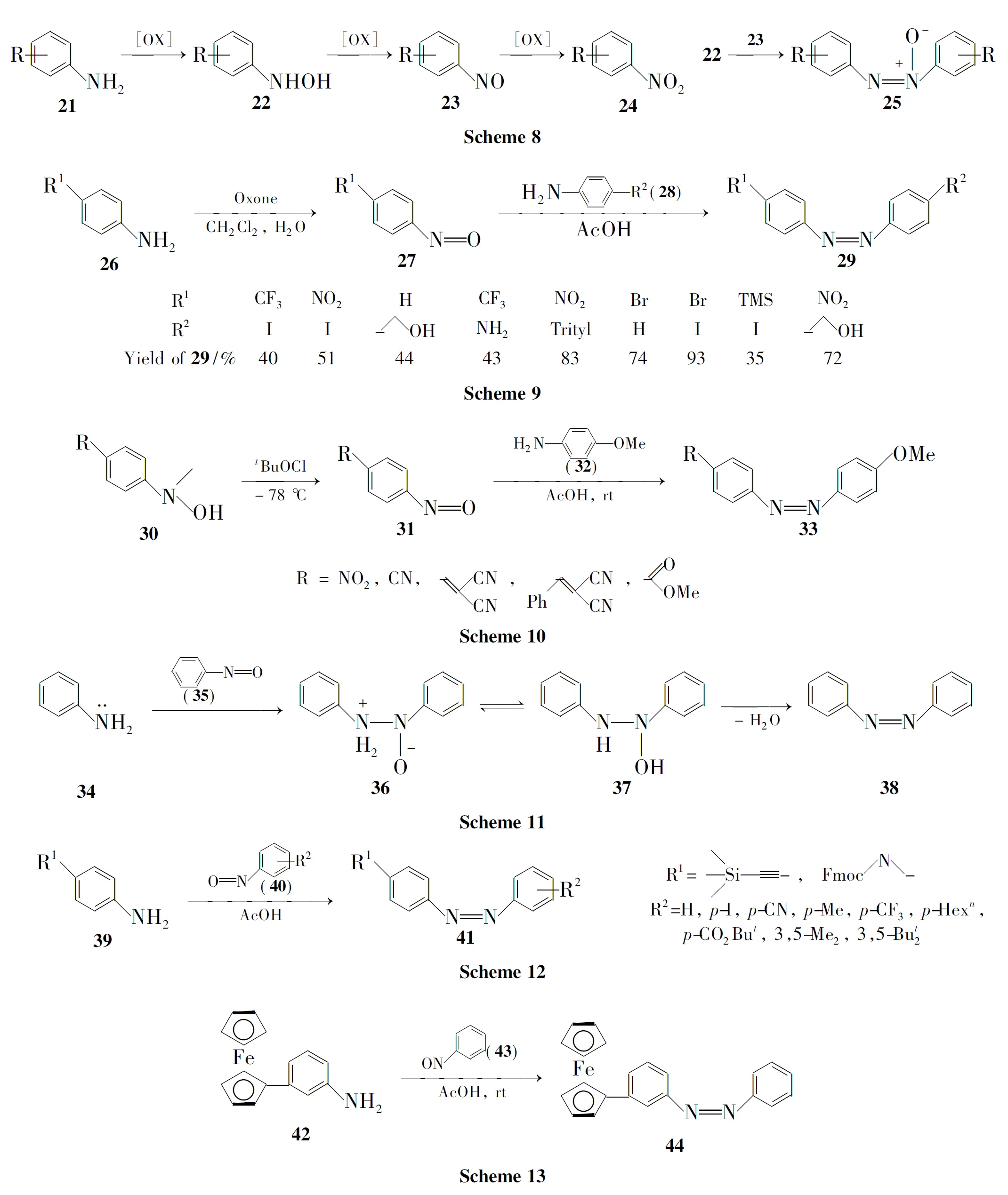
如2010年,Herges R等[28]先用金属锌粉在碱性条件下把芳香硝基化合物还原成为相应的亚硝基化合物(40),再与芳香胺(39)反应制备了一系列偶氮衍生物(41,Scheme 12);Satamoto A等[29]报道了二茂铁偶氮化合物的合成。他们将二茂铁芳香胺(42)和对亚硝基苯甲酸(43)在乙酸中反应制得偶氮化合物(44,Scheme 13),产率24%。
1.3 胺的氧化
胺的氧化法通常制得对称的芳香偶氮化合物。常用的氧化剂有MnO2,KMnO4,H3BO3和HgO/I2等。1999年,Noureldin N A等[30]研究了吸附在五水硫酸铜上的KMnO4的氧化作用。该氧化反应优点多、产率高、反应过程简单、氧化剂安全经济易得。即芳香胺(45)在CH2Cl2溶剂中氧化合成了一系列偶氮苯化合物(46,Scheme 14)。

鉴于有机溶剂对环境的危害,研究者们致力于无溶剂体系的研究。Shaabani A等[31]研究了无溶剂体系中KMnO4的氧化,将芳香胺(47)在Al2O3和CuSO4·5H2O存在下与KMnO4反应制得相应的偶氮化合物(48,Scheme 15),产率较高。
对于H3BO3的氧化作用,Naeimi H等[32]报道了4,4′-二氨基偶氮苯(50)的合成。在四水过硼酸钠,硼酸及醋酸的作用下,将对氨基乙酰苯胺(49)氧化,然后用盐酸,甲醇进行后处理得50(Scheme 16),总产率52%。
Orito K等[33]则将芳香胺(51)在HgO-I2的氧化作用下反应合成偶氮化合物(52,Scheme 17),产率87%。但是该方法的缺点是反应中HgO和I2需过量,会生成对环境有害的重金属盐HgI2。该作者还对反应机理进行了深入研究[33],认为HgO-I2与芳香胺的N原子相互作用形成中间体(53),通过单电子转移形成阳离子(54或55),接着N-N偶联形成化合物(56);HgO-I2与56的N原子相互作用形成中间体57,再经一个双电子氧化过程,脱水形成芳香偶氮苯(38,Scheme 18)。
研究者们也相继开发了一些新的金属和非金属氧化剂,如AgCO3[34],AgO[35],AgMnO4[36],MnO2[37],KO2[38],NaBO3[39],Pb(OAc)4[40],BaMnO4[41],Ce(OH)3O2H[42],镍过氧化物[43],RuCl3/H2O2[44]及高价碘化物如[PhI(OAc)2][45]等;还有以氧气作为氧化剂的反应体系,如O2-KO-tBu[46],O2/Co3O4[47],过氧化物酶/H2O2[48]和O2/CuCl[49]等。
2012年,Youhei T等[50]用有机氧化剂tBuOI氧化芳香胺(58)得偶氮苯(59,Scheme 19)。该反应对底物的适用范围更广,可合成非对称的偶氮化合物(62,Scheme 20),并对杂环偶氮化合物的合成进行了尝试。作者还对该反应的机理进行了深入研究,认为芳香胺的N原子首先在tBuOI的作用下发生第一个碘代过程——NH2和tBuOI发生反应形成中间体(64);64会迅速转化成一碘代铵盐(65);接着通过去质子化作用形成一碘代芳香胺(66),同时生成副产物tBuOH。然后,经历一个相同的碘代过程,生成二碘代芳香胺(67)。值得注意的是,在碘代过程中,控制反应的决定因素不是N-H键的酸度,而是芳香胺的亲核性,亲核性越强,越有利于形成氮卤键(N-I),进而发生之后的反应。因此,带有供电子基团(EDG)的芳香胺更容易发生这个反应。最后带有吸电子基团(EWG)的芳香胺(68)进攻67的N原子,使N-N键偶联,形成化合物69,脱去HI后形成芳香偶氮化合物(70,Scheme 21)。
该反应虽有很多突破,但缺点是所使用的tBuOI具有一定的危险性,容易爆炸;反应需要惰性气体保护;为了防止tBuOI分解,溶剂需除水。


近年来,胺的催化氧化反应不断发展。2008年,Grirrane A等[51]用负载在TiO2上的Au纳米粒子作为催化剂,采用O2氧化芳香胺,合成了一系列偶氮化合物(Scheme 22,Scheme 23)。
该反应开创了催化氧化芳香胺合成偶氮化合物的新局面,具有诸多优点:(1)较高的转化率和选择性;(2)合成了非对称偶氮化合物;(3)氧化剂为O2,符合绿色化学的要求。作者还研究了硝基苯生成偶氮苯的反应,在Au纳米粒子催化下,先将硝基苯还原成氨基苯,再采用O2氧化,生成偶氮苯(Scheme 24)。
作者还对反应的机理进行了深入研究,认为在Au+的作用下,芳香胺经单电子氧化作用形成自由基正离子(54),与中性的胺进行偶合,形成一个含有N-N三电子σ键的化合物(77);77相继失去两个质子,一个电子生成化合物56,在Au+的作用下,56经过单电子氧化作用形成自由基正离子(79),接着失去两个质子和一个电子后生成偶氮苯(38,Scheme 25)。由反应机理可以看出,若某种物质含有较多空位缺陷,可以吸收电子,则可促进该反应的进行,如该反应中的TiO2[51]。
对于上述反应,缺点在于Au纳米粒子造价昂贵,工艺复杂;反应条件较为苛刻:压力高(0.5MPa),温度高(100℃)。为了改进这些缺点,2010年,Zhang C等[52]用铜盐做催化剂,以吡啶做配体,在甲苯中通O2或空气,在较低温度和压力下合成了对称和非对称的偶氮化合物(Scheme 26和Scheme 27),且该反应对取代基的耐受性更强。
该反应的机理(Scheme 28)与之前反应类似,均形成了含有N-N三电子σ键的中间体,不同之处在于参与电子转移的不是Au+,而是配合物Ⅰ。该配合物反应活性很高,可以保证反应的顺利进行[52]。
虽然反应有了很大改进,但产率仍然有待提高。2013年,Zhu Y G等[53]以相同的金属盐-CuBr为催化剂,选用氮代二叔丁基二氮杂环丙酮做氧化剂,合成了一系列偶氮化合物(91,Scheme 29)。该反应产率大大提高,最高可达98%,且氧化剂经济易得,不用惰性气体保护。同时反应对取代基的位置、电子效应没有限制。研究者还发现用该方法氧化芳香亚胺可以合成一系列芳肼类化合物。
为了开发新的金属纳米粒子催化剂,2013年,Cai S F等[54]利用单分散的金属纳米粒子,包括Ag,Pt,Co,Cu,Ni,Pd和Au,成功氧化芳香胺合成了偶氮化合物(93,Scheme 30),其中Ag纳米粒子的反应活性最高,并且反应条件温和,在大气压下即可完成,底物适用范围广泛。
该作者对纳米粒子粒径对反应的影响进行了研究,发现比表面积越大,催化能力越强。该反应的机理如Scheme 31所示。与之前反应类似,先形成芳香胺自由基阳离子(54),然后形成N-N三电子σ键的中间体77,最后氧化生成偶氮苯(Scheme 31)。不同之处在于,该反应形成了过氧离子,参与电子转移。由ESP曲线观测到了过氧离子和胺自由基阳离子的信号,验证了反应机理的可行性。
虽然催化反应大大促进了该类反应的发展,具有很多优点,但是仍然存在不足,如纳米粒子制备过程复杂,某些反应条件较为苛刻,需要高温高压等,新的合成方法仍有待研究。
2007年,本课题组[55]用4-氨基-1,2,4-三唑在氧化剂二氯异腈尿酸钠(SDIC)的作用下合成了偶氮化合物(95和96,Scheme 32)。并于2010年对95的性能进行了实际测试和理论计算[56],研究表明该化合物具有密度高、热稳定性好、能量高等优点,是性能优异的含能化合物。
为了发展这种方法,2010年,本课题组[57]又以1-氨基-1,2,3-三唑为底物,在SDIC作用下合成了偶氮化合物(98,Scheme 33)。该化合物不仅热稳定性好,而且具有光致变色性,可应用于光信息存储、光调控、光开关、光学器件材料等领域。
运用相似的方法,2011年,Thomas等[58]将1-氨基四唑进行偶联得偶氮化合物(100,Scheme 34)。100生成热很高,但热稳定较差,也具有光致变色性。
由上可以看出,SDIC法合成芳香偶氮化合物具有诸多优点:(1)在常温常压下进行,无需惰性气体保护;(2)反应过程简单易控制,产率高;(3)更重要的是该反应对连在N原子上的氨基进行了氧化偶联。若能发展成为一种合成方法,将有广阔的应用前景。


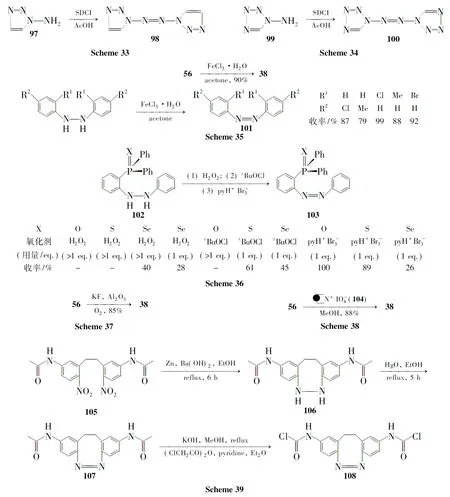
1.4 芳肼的的氧化
1999年,Wang C L等[59]用FeCl3在丙酮中氧化芳肼56得偶氮苯38(Scheme 35),产率90%。用这种氧化方法,还可合成化合物101(Scheme 35),产率79~99%。

为了响应绿色化学的发展,科学家始终致力于无溶剂反应体系的发展。2007年,Mihara M等[61]在无溶剂体系中用KF/Al2O3氧化芳肼56合成偶氮苯38(Scheme 37),产率85%。
为了提高反应产率,简化后处理步骤,该反应还可以采用固相反应,在树脂高碘酸盐的氧化下进行。Barth M等[62]用与树脂反应制得的高碘酸盐(104)氧化56合成偶氮苯38(Scheme 38),产率88%左右。
除此之外,上述反应还可以用NH4VO3[63],CuCl2[64],Co(Ⅱ)络合物[65-66]和FeSO4/KClO3/H2SO4[67]体系氧化。
近来,Kubitschke J等[68]将硝基化合物(105)还原合成桥环芳肼(106),再采用HgO在乙醇中氧化106得桥环偶氮化合物(107),作为目标产物(108)的前体原料(Scheme 39)。
1.5 硝基化合物的还原偶联反应
该反应可使用的还原剂有很多,如LiAlH4[69],NaBH4[70],KOH[71],Zn/NaOH[72],Bi[73],Bi-KOH[74]和Pb/HCO2NH4[75]等,是获得对称芳香偶氮化合物的有效方法。
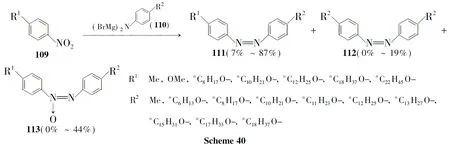
在1989年,Okubo M等[76]就发现硝基芳香衍生物(109)在试剂(110)的作用下,可生成不对称的偶氮化合物(111),对称的偶氮化合物(112)和氧化偶氮化合物(113,Scheme 40)。由于缺乏选择性,该反应的应用受到了限制。
为了避免反应中出现重排异构化副反应,Srinivasa G R等[77]利用催化转移加氢方法,采用HCO2HNEt3/Mg还原体系还原硝基芳香苯(114),高产率合成了一系列芳香偶氮衍生物(115,Scheme 41)。在该反应中,三乙基甲酸铵作为氢的给予体,定量释放氢,而催化剂Mg则作为氢的转移剂将氢转移给反应底物,反应底物接受氢后被还原。该反应快速,条件温和,在室温下就可以发生,并且产率很高。该课题组还用Pb代替Mg进行尝试合成了一系列偶氮衍生物,最高产率可达92%,并且Pb可以回收再利用[78]。
还有很多还原体系可以用于硝基芳香化合物的还原偶联反应。例如使用超声环境[79],SnCl2/NaOH[80],Al/NaOH[81],Pb/HCO2HNEt3[82],Pb/CH3CO2NH4[83],TiCl4/LiAlH4[84],NaBH4/(PhTe)2[85]和Co(CO)8[86]等,生成的偶氮化合物产率60%~95%。
这类反应很少用到催化体系。但2010年,Zhu H等人[87]用负载在ZrO2上的Au纳米粒子作催化剂,在可见光或紫外光的作用下,还原硝基苯衍生物(116),生成了相应的偶氮化合物(117,Scheme 42)。该方法具有很高的选择性,且反应条件温和。
2013年,Wang J Q等[88]用Pd(acac)2等金属盐作催化剂,用H2作还原剂,超高产率合成了芳香偶氮化合物(119,Scheme 43)。在反应过程中可原位生成Pb纳米粒子,对反应起催化作用。经研究,该反应不仅省去了纳米粒子的制备步骤,可以一步反应,而且原位生成的Pb纳米粒子比单独合成的纳米粒子催化效率更高。此外,该反应对基团的耐受性很强,对取代基的电子效应,取代位置有很大的包容性。
该反应的机理如Scheme 44所示,金属盐Pd(acac)2在H2作用下原位生成Pd纳米粒子,芳香硝基苯76在纳米粒子的催化下,生成中间体(120),脱水后形成氧化偶氮苯(121),被H2还原即可生成偶氮苯[88](Scheme 44)。
1.6 重氮盐的二聚反应
当Cu和酸同时存在时(Gatterman方法),或者在铜盐的作用下,重氮盐就容易发生二聚作用生成芳香偶氮化合物[89]。
该反应在很大程度上受芳环取代基性质的影响。当芳环上含有吸电子基时,如化合物(122),容易发生C-C偶联而主要生成联苯化合物(123),而当取代基为供电子基时,则容易生成偶氮化合物(124,Scheme 45)。经研究,当Cu2+和重氮盐的浓度升高,Cu+浓度降低时,偶氮化合物的比例会升高[90]。

1.7 叠氮化合物的热解
在芳香胺存在下,加热叠氮化合物会生成不对称偶氮化合物,但产率较低。如4-硝基叠氮苯(125)在4-甲基胺(126)的存在下,加热至135℃会生成偶氮苯衍生物(127,Scheme 46),产率16%[91]。当两个芳香环取代基的电子效应不同时,该反应更容易发生。叠氮化合物不稳定,容易爆炸,限制了该类反应的发展。
1.8 其他方法
芳香氧化偶氮化合物在硫酸作用下,重排为对位为酚羟基取代的偶氮化合物,该反应称为Wallach重排反应。如1983年,Shimao I等[92]进行了如下反应,氧化偶氮苯衍生物(128)在H2SO4存在下加热至100℃生成两种羟基偶氮化合物(129和130)和一种偶氮苯衍生物(131)。4-硝基衍生物主要生成129,当取代基为COCH3和COOH时,主要产物为130(Scheme 47)。
缩醛和芳肼之间反应也可生成芳香偶氮化合物。2004年,Carreno M C等[93]报道了缩醛132和芳肼133反应可以生成芳香偶氮苯(134,Scheme 48)。加入催化剂量的(NH4)2[Ce(NO3)6](即CAN)可以缩短反应时间。
醌类化合物和芳肼反应也可以生成偶氮化合物。如2,6-二甲基-1,4-苯醌(135)和对硝基苯肼(136)反应可生成偶氮化合物(137,Scheme 49),产率77%[94]。
芳钙衍生物和N2O反应也可以生成偶氮化合物。1995年,Hays M L等[95]将碘苯(138),金属Ca和N2O在二甲氧基乙烷(DME)中反应得偶氮苯38(Scheme 50),产率61%。
此外,2004年,Ziegler T等[96]将苯并三唑类衍生物(139)和酚类化合物(140)反应获得一系列偶氮苯衍生物(141,Scheme 51)。



2 结论
合成偶氮化合物最常用的方法是重氮偶联反应和米尔斯反应。对于重氮偶联反应,主要合成不对称偶氮化合物,该反应有如下缺点:(1)重氮盐有时不稳定,会发生爆炸;(2)要注意控制反应温度和pH,否则会生成较多副产物或者不发生反应;(3)对反应底物有限制。
对于米尔斯反应,需要先制得亚硝基芳香化合物,胺反应活性不高时,会产生比较多的副产物,同时对反应底物也有限制。胺的氧化主要制得对称芳香偶氮化合物,传统的氧化剂中通常含有重金属离子,有害环境,但催化氧化大大提高了该反应的适用性。
硝基芳香化合物的还原偶联反应多获得对称的偶氮化合物,缺点是常用的还原剂中多含有重金属离子,且反应时需过量,由此产生的副产物会污染环境。重氮盐的二聚反应,只对个别反应底物有作用,不能发展为一种合成方法。由于多数叠氮化合物不稳定,易爆炸,叠氮化合物热解生成偶氮化合物的反应也由此受到了限制。
综上所述,现有的合成方法多存在一些不足之处,总体来讲,(1)反应底物有限,主要针对苯环,极少针对杂环;(2)多数反应只针对芳香环的C-NH2进行偶联,没有针对环上的N-NH2进行偶联;(3)反应多使用有机溶剂,且还原剂、氧化剂多含有重金属离子,不符合绿色化学的要求;(4)反应条件较苛刻,如无氧无水条件,高温高压条件等。所以,新的合成方法还有待研究。特别是更注重研发一些低毒高效又绿色环保的合成策略。总之,芳香偶氮衍生物不仅激发和丰富了化学家的创造欲和想象力,而且还为材料学家、生物学家及药物学家都带来前所未有的创新机遇和发展空间。
[1] Mermut O,Barrett C J.Using light to control physical properties of polymers and surfaces with azobenzene chromophores[J].Pure and Applied Chemistry,2004,76(7-8):1445-1465.
[2] Zollinger H.Colour Chemistry.Syntheses,Properties and Applications of Organic Dyes and Pigments[M].New York:VCH,1987.
[3] Ikeda T,Tsutumi O.Optical switching and image storage by means of azobenzene liquid-crystal films[J].Science,1995,268(5219):1873-1975.
[4] Huynh M H V,Hiskey M A,Hartline E L,etal.Polyazido high-nitrogen compounds:Hydrazo and azo-1,3,5-triazine[J].Angewandte Chemie International Edition,2004,43(37):4924-4928.
[5] Qi C,Li S H,Li Y C,etal.Synthesis and promising properties of a new family of high-nitrogen compounds:Polyazido and polyamino substitutedN,N′-azo-1,2,4-triazoles[J].Chemistry——a European Journal,2012,18(51):16562-16570.
[6] Naud D L,Hiskey M A,Harry H H.Synthesis and explosive properties of 5,5′-dinitro-3,3′-azo-1H-1,2,4-triazole[J].Journal of Energetic Materials,2003,21(1):57-62.
[7] Haghbeen K,Tan E W.Synthesis of catechol azo dyes[J].The Journal of Organic Chemistry,1998,63(19):4503-4505.
[8] Merrington J,James M,Bradley M.Supported diazonium salts-convenient reagents for the combinatorial synthesis of azo dye[J].Chemical Communications,2002,(2):140-141.
[9] 俞凌翀.基础理论有机化学[M].北京:人民教育出版社,1981.
[10] Tsai W H,Shiao Y J,Lin S J,etal.Selective COX-2inhibitors.Part 1:Synthesis and biological evaluation of phenylazobenzenesulfonamides[J].Bioorganic &Medicinal Chemistry Letters,2006,16:4440-4443.
[11] Curtin D V,Tveten J L.Reaction of diarylzine reagents with aryldiazonium salts.Direct formation of cis-azo compounds[J].The Journal of Organic Chemistry,1961,26(6):1764-1768.
[12] Hunter C A,Sarson L D.Synthesis and photochemical properties of covalently-connected azobenzene-porphyrin derivatives[J].Tetrahedron Letters,1996,37(5):699-702.
[13] Hegarty A F.In the chemistry of diazonium and diazo group[M].New York:Wiley,1978.
[14] Zollinger H.Diazo chemistry I.Aromatic and Heteroaromatic Compounds[M].New York:VCH,1994.
[15] Zollinger H.Azo and Diazo Chemistry Aliphatic and Aromatic Compounds[M].New York:Interscience,1961.
[16] Tsuge A,Moriguchi T,Mataka S,etal.Evaluation of through-space interaction in [2.2]metacyclophanes by diazo coupling reaction[J].Journal of the Chemical Society,Perkin Transactions 1,1993,(18):2211-2215.
[17] Kim L K,Kim G,Kim C R,etal.UV band splitting of chromogenic azo-coupled calix[4]crown upon cation complexation[J].The Journal of Organic Chemistry,2003,68(5):1933-1937.
[18] Desroches C,Parola S,Vocanson F,etal.Synthesis,characterization and optical power limiting behaviour of phenylazo- and 4-nitrophenylazo-tetrahydroxy tetrathiacalix[4]arene[J].Journal of Materials Chemistry,2001,11(12):3014-3017.
[19] Lhotak P,Moravec J,Stibor I.Diazo coupling:An alternative method for the upper rim amination of thiacalix[4]arenes[J].Tetrahedron Letters,2002,43(20):3665-3668.
[20] Entwistle I D,Gilkerson T,Johnstone R A W.Rapid catalytic transfer reduction of aromatic nitro compounds to hydroxylamines[J].Tetrahedron,1978,34(2):213-215.
[21] Caro H.Hauptversammlung des Vereins deutscher Chemiker am 1.bis 4.Juni 1898zu Darmstadt[J].Angewandte Chemie,1898,11(36):845.
[22] Ibne-Rasa K M,Lauro C G,Edwards J O.Mechanism of the oxidation of nitrosobenzenes by peroxoacetic acid[J].Journal of the American Chemical Society,1963,85(8):1165-1167.
[23] Bleasdale C,Ellis M K,Farmer P B,etal.Synthesis and spectroscopic characterisation of 3-chloroperbenzoic acid-17O,18O,nitrosobenzene-17O,18O and nitrosobenzene-15N[J].Journal of Labelled Compounds and Radiopharmaceuticals,1993,33(8):739-746.
[24] Brill E.Diethyl azodicarboxylate oxidation of some carcinogenic arylhydroxylamines to nitroso derivatives[J].Experientia,1969,25(7):680.
[25] Berry O J,Collins I,Roberts S M,etal.Polychloroaromatic compounds.Part V.Preparation and oxidation of pentachlorophenyl-substituted tertiary amines and reactions ofn-butyl-lithium and other nucleophiles with various pentachlorophenyl derivatives[J].Journal of the Chemical Society C,1969,(9):1285-1294.
[26] Yu B C,Shirai Y,Tour J M.Syntheses of new functionalized azobenzenes for potential molecular electronic devices[J].Tetrahedron,2006,62(44):10303-10310.
[27] Davey M H,Lee V Y,Marks T J,etal.Synthesis of aryl nitroso derivatives by tert-butyl hypochlorite oxidation in homogeneous media.Intermediates for the preparation of high-hyperpolarizability chromophore skeletons[J].The Journal of Organic Chemistry,1999,64(13):4976-4979.
[28] Kubitschke J,Näther C,Herges R.Synthesis of functionalized triazatriangulenes for application in photo-switchable self-assembled monolayers[J].European Journal of Organic Chemistry,2010,(26):5041-5055.
[29] Sakamoto A,Hirooka A,Namiki K,etal.Photon-,electron-,and proton-induced isomerization behavior of ferrocenylazobenzenes[J].Inorganic Chemistry,2005,44(21):7547-7558.
[30] Noureldin N A,Bellegarde J W.A novel method.The synthesis of ketones and azobenzenes using supported permanganate[J].Synthesis,1999,(6):939-942.
[31] Shaabani A,Lee D G.Solvent free permanganate oxidations[J].Tetrahedron Letters,2001,42(34):5833-5836.
[32] Naeimi H,Safari J,Heidarnezhad A.Synthesis of Schiff base ligands derived from condensation of salicylaldehyde derivatives and synthetic diamin[J].Dyes and Pigments,2007,73(2):251-253.
[33] Orito K,Hatakeyama T,Takeo M,etal.Dimerization of anilines and benzylamines with mercury(Ⅱ)oxide-iodine reagent[J].Tetrahedron,1998,54(29):8403-8410.
[34] Fetizon M,Golfier M,Milcent R,etal.Oxydations par le carbonate d'argent-Ⅻ:Groupes fonctionnels azotes[J].Tetrahedron,1975,31(2):165-170.
[35] Ortiz B,Villanueva P,Walls F.Silver(Ⅱ)oxide as a reagent.Reactions with aromatic amines and miscellaneous related compounds[J].The Journal of Organic Chemistry,1972,37(17):2748-2750.
[36] Firouzabadi H,Vessal B,Naderi M.Bispyridinesilver permanganate[Ag(C5H5N)2]MnO4:An efficient oxidizing reagent for organic substrates[J].Tetrahedron Letters,1982,23(17):1847-1850.
[37] Hombrecher H K,Ludtke K.Synthesis and spectroscopic investigation of directly azobenzene bridged diporphyrins[J].Tetrahedron,1993,49(42):9489-9494.
[38] Crank G,Makin M I H.Oxidations of aromatic amines by superoxide ion[J].Australian Journal of Chemistry,1984,37(4):845-855.
[39] Mehta S M,Vakilwala M V.Sodium perborate as a reagent in organic chemistry.Ⅰ.Preparation of azo-compounds[J].Journal of American Chemistry Society,1952,74(2):563-564.
[40] Baer E,Tosoni A L.Formation of symmetric azo-compounds from primary aromatic amines by lead tetraacetate[J].Journal of American Chemistry Society,1956,78(12):2857-2858.
[41] Firouzabadi H,Mostafavipoor Z.Barium manganate.A versatile oxidant in organic synthesis[J].Bulletin of the Chemical Society of Japan,1983,56(3):914-917.
[42] Firouzabadi H,Mostafavipoor Z.Ceric trihydroxy hydroperoxide Ce(OH)3O2H,a regenerable,mild,and a versatile reagent for the oxidation of organic compounds[J].Synthetic Communications,1984,14(9):875-882.
[43] Nakagawa K,Tsuji T.Oxidation with nickel peroxide.Ⅱ.Oxidation of amines[J].Chemical and Pharmaceutical Bulletin,1963,11(3):296-301.
[44] Barak G,Sasson Y.Effect of phase-transfer catalysis on the selectivity of hydrogen peroxide oxidation of aniline[J].The Journal of Organic Chemistry,1989,54(14):3484-3486.
[45] Pausacker K H.Oxidations with phenyl iodosoacetate.Part Ⅱ.The oxidation of primary aromatic amines[J].Journal of the Chemical Society,1953,(0):1989-1990.
[46] Horner L,Dehnert J.Azo-aryle und phenazine aus primären arylaminanionen durch autoxydation[J].Chemische Berichte,1963,96(3):786-797.
[47] Belew J S,Garza C,Mathieson J M.Catalysis:Autoxidation in the presence of active cobalt oxide[J].Journal of the Chemical Society D,Chemical Communications,1970,(11):634-635.
[48] Hughes G M K,Saunders B C.Studies in peroxidase action.Part Ⅸ.Reactions involving the rupture of the C-F,C-Br,and C-I links in aromatic amines[J].Journal of the Chemical Society,1954,4630-4634.
[49] Kinoshita K.On the mechanism of oxidation by cuprous chloride,pyridine and air.Ⅰ.The properties of the reaction[J].Bulletin of the Chemical Society of Japan,1959,32(8):777-780.
[50] Youhei T,Sota O,Satoshi M.Oxidative dimerization of aromatic amines usingt-BuOI:Entry to unsymmetric aromatic azo compounds[J].Angewandte Chemie,2012,124(31):7924-7928.
[51] Grirrane A,Corma A,Garcia H.Gold-catalyzed synthesis of aromatic azo compounds from anilines and nitroaromatic[J].Science,2008,322(5908):1661-1664.
[52] Zhang C,Jiao N.Copper-catalyzed aerobic oxidative dehydrogenative coupling of anilines leading to aromatic azo compounds using dioxygen as an oxidant[J].Angewandte Chemie International Edition,2010,49(35):6174-6177.
[53] Zhu Y G,Shi Y.Facile Cu(Ⅰ)-catalyzed oxidative coupling of anilines to azo compounds and hydrazines with diaziridinone under mild conditions[J].Organic Letters,2013,15(8):1942-1945.
[54] Cai S F,Rong H P,Yu X F,etal.Room temperature activation of oxygen by monodispersed metal nanoparticles:Oxidative dehydrogenative coupling of anilines for azobenzene syntheses[J].ACS Catalysis,2013,3(4):478-486.
[55] Li S H,Pang S P,Li X T,etal.Synthesis of new tetrazene(N-N=N-N)-linked bi(1,2,4-triazole)[J].Chinese Chemical Letters,2007,18:1176-1178.
[56] Qi C,Li S H,Wang Y,etal.A novel stable high-nitrogen energetic material:4,4′-azobis (1,2,4-triazole)[J].Journals of Materials Chemistry,2011,21:3221-3225.
[57] Li Y C,Qi C,Li S H,etal.1,1′-Azobis-1,2,3-triazole:A high-nitrogen compound with stable N8structure and photochromism[J].Journal of the American Chemical Society,2010,132(35):12172-12173.
[58] Thomas M K,Davin G P.1,1′-Azobis(tetrazole):A highly energetic nitrogen-rich compound with a N10chain[J].Inorganic Chemistry,2011,50(7):2732-2734.
[59] Wang C L,Wang X X,Wang X Y,etal.A convenient method for dehydrogenation of symmetric hydrazo compounds[J].Synthetic Communications,1999,29(19):3435-3438.
[60] Yamamura M,Kano N,Kawashima T.Systematic study on the structures and reactivity of hydrazobenzenes and azobenzenes bearing a chalcogenophosphoryl group[J].Inorganic Chemistry,2006,45(16):6497-6507.
[61] Mihara M,Nakai T,Iwai T,etal.Solvent-free basic or KF/alumina-assisted dehydrogenation of hydrazo compounds[J].Synlett,2007,13:2124-2126.
[62] Barth M,Tasadaque S,Shah A,etal.High loading polymer reagents based on polycationic Ultraresins.Polymer-supported reductions and oxidations with increased efficiency[J].Tetrahedron,2004,60(39):8703-8709.
[63] Trischler F J.The determination of hydrazobenzene by the die method with hydrogen peroxide catalyzed with vanadium[J].Journal of Thermal Analysis and Calorimetry,1979,16(1):119-122.
[64] Blackadder D A,Hinshelwood C.The kinetics of the rearrangement and oxidation of hydrazobenzene in solution.Part Ⅱ.The catalysed oxidation[J].Journal of the Chemical Society,1957,2904-2906.
[65] Nemeth S,Szeverenyi Z,Simandi L I.Catalytic oxidations with molecular oxygen in the presence of cobaloxime(Ⅱ)derivatives[J].Inorganica Chimica Acta,1980,44:L107-L109.
[66] Kim S S B,Hommer R B,Cannon R D.The oxidation of hydrazobenzene catalyzed by cobalt complexes in nonaqueous solvents[J].Bulletin of the Korean Chemical Society,2006,27(2):255-265.
[67] Shi L,Pan F,Jia X S,etal.A simple,efficient and new catalyst oxidation method with KClO3/H2SO4/FeSO4for preparing bisazo compounds[J].Synthetic Communications,2001,31(11):1691-1695.
[68] Samanta S,Qin C G,Lough J A,etal.Bidirectional photocontrol of peptide conformation with a bridged azobenzene derivative[J].Angewandte Chemie International Edition,2012,51(26):6452-6455.
[69] Nystrom R F,Brown W G.Reduction of organic compounds by lithium aluminum hydride.Ⅲ.Halides,quinones,miscellaneous nitrogen compounds[J].Journal of the American Chemical Society,1948,70(11):3738.
[70] Hutchins R O,Lamson D W,Rua L,etal.Reduction of aromatic nitro compounds with sodium borohydride in dimethyl sulfoxide or sulfolane.Synthesis of azo or azoxy derivatives[J].The Journal of Organic Chemistry,1971,36(6):803-806.
[71] Wei W H,Tomohiro T,Kodaka M,etal.Selective synthesis and kinetic measurement of 1∶1and 2∶2cyclic compounds containing 1,4,7,10-tetraazacyclododecane and azobenzene units[J].The Journal of Organic Chemistry,2000,65(26):8979-8987.
[72] Khan A,Hecht S.Towards photocontrol over the helix-coil transition in foldamers: synthesis and photoresponsive behavior of azobenzene-core amphiphilic oligo(meta-phenylene ethynylene)s[J].Chemistry——A European Journal,2006,12(18):4764-4774.
[73] Wada S,Urano M,Suzuki H.The newborn surface of dull metals in organic synthesis.Bismuth-mediated solvent-free one-step conversion of nitroarenes to azoxy- and azoarenes[J].The Journal of Organic Chemistry,2002,67(23):8254-8257.
[74] Laskar D D,Prajapati D,Shandu J S.Bi-KOH.An efficient reagent for the coupling of nitroarenes to azo and azoxy compounds[J].Journal of the Chemical Society,Perkin Transactions 1,2000,(1):67-69.
[75] Gowda S,Gowda D C.Application of lead and ammonium formate as a new system for the synthesis of azo compounds[J].Synthesis,2002,(4):460-462.
[76] Okubo M,Matsuo K,Yamauchi A.Aryliminodimagnesium reagents.XV.Condensation with nitrobenzene.Formation of unsymmetrical azobenzenes favored by long-chainp-alkoxy-substituted reagents[J].Bulletin of the Chemical Society of Japan,1989,62(3):915-918.
[77] Srinivasa G R,Abiraj K,Gowda D C.Facile synthesis of azo compounds from aromatic nitro compounds using magnesium and triethylammonium format[J].Australian Journal of Chemistry,2004,57:609-610.
[78] Srinivasa G R,Abiraj K,Gowda D C.The synthesis of azo compounds from nitro compounds using lead and triethylammonium formate[J].Tetrahedron Letters,2003,44(31):5835-5837.
[79] Pasha M A,Jayashankara V P.Reduction of arylnitro compounds to azoarenes and/or arylamines by Al/NaOH in methanol under ultrasonic conditions[J].Ultrasonics Sonochemistry,2005,12(6):433-435.
[80] Krzysztof S,Renata B,Anna S,etal.Functionalization of electrode surfaces with monolayers of azocompounds and gold clusters[J].Journal of Inclusion Phenomena and Macrocyclic Chemistry,2004,49(1-2):173-179.
[81] Ameerunisha S,Zacharias P S.Characterization of simple photoresponsive systems and their applications to metal ion transport[J].Journal of the Chemical Society,Perkin Transactions 2,1995,(8):1679-1682.
[82] Srinivasa G R,Abiraj K,Gowda D C.The synthesis of azo compounds from nitro compounds using lead and triethylammonium formate[J].Tetrahedron Letters,2003,44(31):5835-5837.
[83] Srinivasa G R,Abiraj K,Gowda D C.Lead-catalyzed synthesis of azo compounds by ammonium acetate reduction of aromatic nitro compounds[J].Synthetic Communications,2003,33(24):4221.
[84] Malinowski M,Kaczmarek L,Rozploch F.Reductive properties of low-valent titanium reagents[J].Journal of the Chemical Society,Perkin Transactions 2,1991,(6):879-883.
[85] Ohe K,Uemura S,Sugita N,etal.Sodium arenetellurolate-catalyzed selective conversion of nitro aromatics to aromatic azoxy or azo compounds and its application for facile preparation of 3,3′- and 4,4′-bis[.beta.-(aryltelluro)vinyl] azobenzenes from (3- and 4-nitrophenyl)acetylenes[J].The Journal of Organic Chemistry,1989,54(17):4169-4174.
[86] Alper H,Paik H N.A convenient synthesis of azobenzenes.Interesting solvent effectss[J].Journal of Organometallic Chemistry,1978,144(1):C18-C20.
[87] Zhu H,Ke X,Yang X,etal.Reduction of nitroaromatic compounds on supported gold nanoparticles by visible and ultraviolet light[J].Angewante Chemie International Edition,2010,49(50):9657-9661.
[88] Wang J Q,Hu L,Cao X Q,etal.Catalysis by Pd nanoclusters generated in situ of high-efficiency synthesis of aromatic azo compounds from nitroaromatics under H2atmosphere[J].RSC Advances,2013,3(15):4899-4902.
[89] Cohen T,Lewarchik R J,Tarino J Z.Role of radical and organocopper intermediates in aromatic diazonium decomposition induced by cuprous ion[J].Journal of the American Chemistry,1974,96(25):7753-7760.
[90] Smith M B,March J.Advanced Organic Chemistry[M].Americal:John Wiley &Sons,5th edn,2001.
[91] Scriven E F V,Suschitzky H.Thermolysis of aryl azides in anilines[J].Tetrahedron Letters,1973,14(2):103-106.
[92] Shimao I,Oae S.The Wallach rearrangement of some 4,4′-disubstituted azoxybenzenes[J].Bulletin of the Chemical Society of Japan,1983,56(2):643-644.
[93] Carreno M C,Mudarra F G,Merino E,etal.Synthesis of azobenzenes from quinone acetals and arylhydrazines[J].The Journal of Organic Chemistry,2004,69(10):3413-3416.
[94] Smith L I,Irvine W B.The structures of arylhydrazones of unsymmetrically substituted quinones[J].Journal of the American Chemistry Society,1941,63(4):1036-1043.
[95] Hays M L,Hanusa T P.A reinvestigation of the reaction of arylcalcium iodides with nitrous oxide[J].Tetrahedron Letters,1995,36(14):2435-2436.
[96] Mico X A,Ziegler T,Subramanian L R.A versatile direct approach to ortho-substituted azobenzenes from benzotriazoles[J].Angewandte Chemie International Edition,2004,43(11):1400-1403.
ResearchProgressontheSynthesisofAromaticAzoCompounds
ZHANG Lu-bo, ZHAO Xiu-xiu, PANG Si-ping, LI Yu-chuan, SUN Cheng-hui, LI Sheng-hua
(School of Materials,Beijing Institute of Technology,Beijing 100081,China)
The main synthetic methods of aromatic azo compounds including the azo coupling reaction,the Mills reaction,the oxidation of anilines,the oxidation of arylhydrazines,the reductive coupling of aromatic nitro derivatives,the dimerization reaction of diazonium salts,the thermolysis of azides and so on,were reviewed with 96references.The advantages and disadvantages of the methods were briefly summarized.
aromatic azo compound;synthetic method;coupling;review
2014-01-10;
2014-08-11
爆炸科学与技术国家重点实验室重点基金资助项目(ZDKT12-03)
张鲁博(1989-),女,汉族,山东聊城人,硕士研究生,主要从事含能材料的合成及性能研究。E-mail: xiaoahexiaob@163.com
李生华,博士,讲师,硕士生导师,E-mail: lishenghua@bit.edu.cn
O625.6
A
1005-1511(2014)05-0709-16
猜你喜欢
成都大学学报(自然科学版)(2021年1期)2021-05-22
成都信息工程大学学报(2019年1期)2019-05-20
心肺血管病杂志(2019年1期)2019-04-22
分析测试学报(2018年12期)2018-12-19
石油化工(2018年6期)2018-07-04
当代化工研究(2016年9期)2016-03-20
中国粮油学报(2016年1期)2016-02-06
化工进展(2015年6期)2015-11-13
中国塑料(2015年8期)2015-10-14
河南科技(2014年16期)2014-02-27
