
IAD法在合成β-甘露糖苷键中的应用
2014-08-08 07:08:20李忆雪
重庆工商大学学报(自然科学版) 2014年12期
李忆雪, 孙 斌
(重庆工商大学 催化与功能有机分子重庆市重点实验室,重庆 400067)
碳水化合物( 糖类) 是自然界中存在最为广泛的生物分子, 几乎存在于所有生命体中。它们通常以相互间的同聚物或杂聚物形式存在, 或者与其他生物大分子, 如脂类或蛋白质, 形成糖复合物而存在于生命体中。在天然多聚糖中,β-吡喃甘露糖非常常见。这些天然多糖包括蛋白聚糖、糖脂、糖蛋白、微生物多糖等一些具有生物活性的天然物质。在糖苷化反应中,通过2-位上的取代基(如乙酰基)的邻基参与效应很容易立体选择性地得到β-甘露糖苷化产物,然而要得到β-甘露糖苷化产物就不是如此简单(图1)[1,2]。虽然控制糖苷化立体选择性的关键因素已基本了解,但是要严格控制形成β-甘露糖苷键仍具挑战性[1,2]。
为了得到β-甘露糖苷键,糖化学家们作了大量探索[3],在这些探索中,分子内糖苷配体转移法(intramolecular aglycon delivery, IAD)最有希望,因为在动力学控制下,它们能预期保证形成β-吡喃甘露糖苷键[4,5]。因此通过分子内糖苷配体转移法的立体选择性糖苷化反应与受体的结构无关。在合成复杂分子时,形成连接臂是实现以区域和立体选择性方式的分子内反应的必要步骤[6,7]。尽管分子内糖苷配体转移法在合成β-甘露糖苷键中的应用已有英文综述[4,8,9],但是还没有相关的中文综述来详细介绍。为此,对分子内糖配体转移法在立体选择性地合成β-甘露糖苷键中的应用进行介绍。
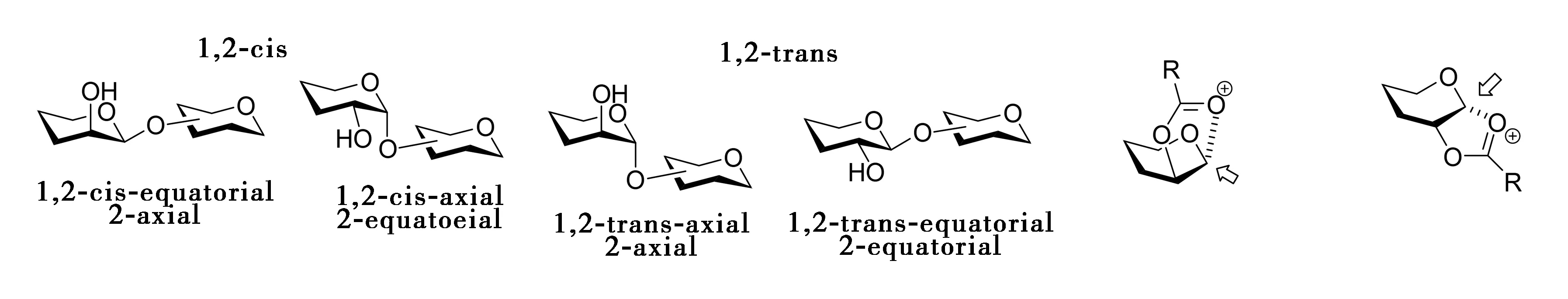
图1 糖苷连接的类型
1 缩酮型连接臂
1.1 二甲基缩酮(异亚丙基缩醛)
选择性地构筑β-连接的甘露糖苷键通常被认为是非常困难的。1991年,Barresi和Hindsgaul率先报道了分子内糖苷配体转移法,这种方法被证明是最有希望能唯一得到β-甘露糖的[10]。他们的策略是首先形成混合二甲基缩酮4作为连接臂中间体。这一步是糖基化受体3与异丙基醚2反应,异丙基醚由乙酸酯1通过Tebbe’s exomethylenation方法(Figure 2)制备[11],接着硫代苷用N-碘代丁二酰亚胺(NIS)活化以高立体控制的方式得到二聚糖5。通过缩酮连接臂固定,保证了以严格动力学方式控制立体定向转移。有趣的是,在糖苷化反应步骤中加入1当量的甲醇,产物中没有发现甲基苷,这说明糖苷化过程是按分子内模式进行的。在糖苷化反应中,加入二叔丁基甲基吡啶(DTBMP)会提高收率,这是因为它的加入抑制了酸性条件下混合缩酮的分解[12](图2)。
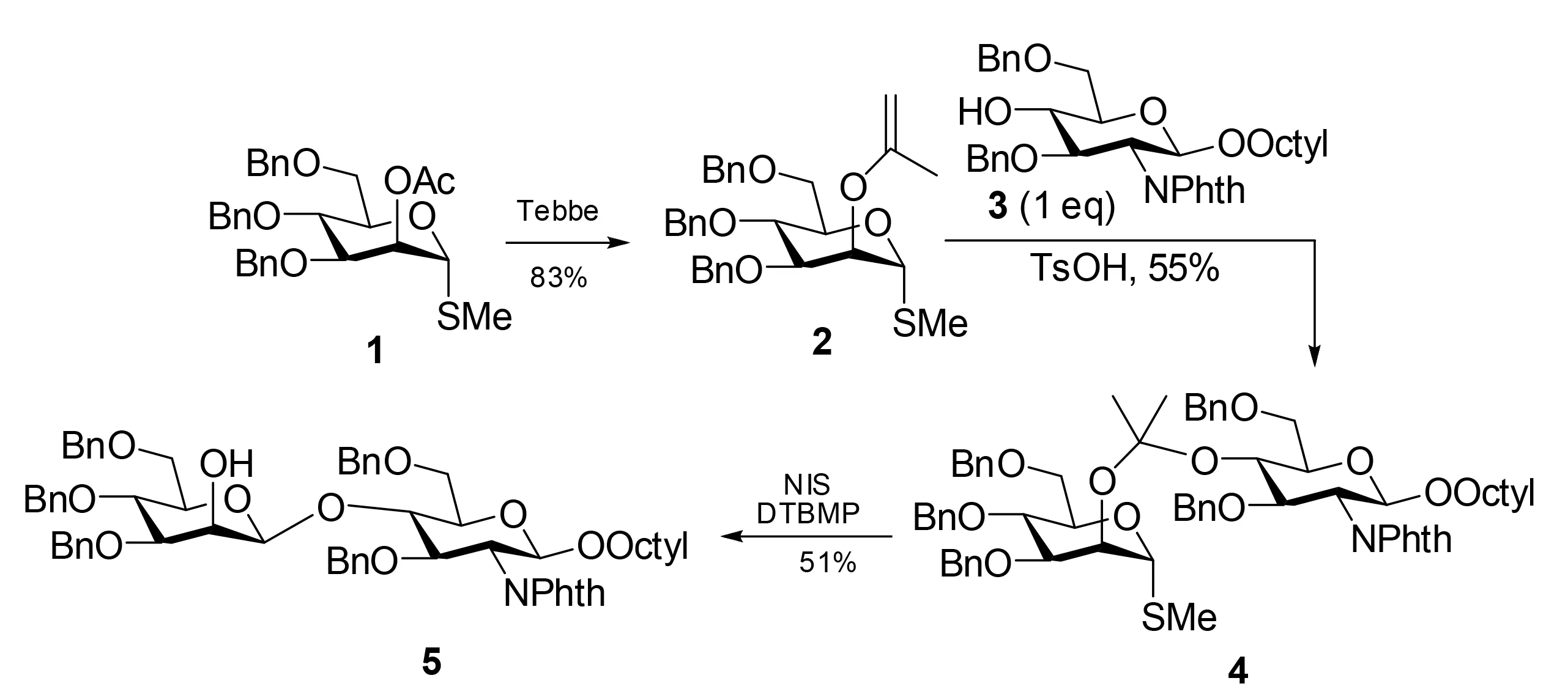
图2 通过二甲基缩酮连接臂中间体的分子内糖苷配体转移
1.2 2-碘甲基-1-甲基和2-碘甲基-1-(4-甲氧苯基)-缩酮
Fairabnks等[13,14]讲述了他们从亚异丙基醚和一系列伯醇合成混合缩酮时遇到的困难,他们发现二者混合会导致亚异丙基醚水解。针对这个问题,他们研究用N-碘代丁二酰亚胺作为一种代替的亲电试剂来促进混合缩醛(缩酮)的形成,即缩酮9由化合物7碘醚化后的产物与化合物8反应后形成。接下来用NIS和DTBMP催化分子内糖苷化反应,立体选择性地得到相应的二聚糖β-Man-(1→6)Gal(10)(图3)。
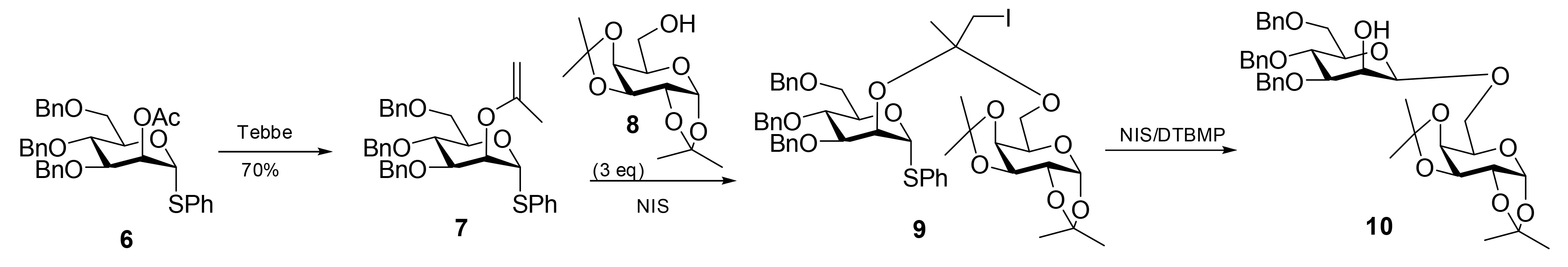
图3 通过2-碘甲基-1-甲基-和2-碘甲基-1-(4-甲氧苯基)-缩酮-连接臂中间体的分子内糖苷配体转移
2 亚烷基缩醛
2.1 2-碘亚丙基缩醛
为了最大限度地提高桥联中间体的形成效率,位阻较小的亚烷基缩醛中间体应用到IAD中,Fairbanks等[15, 16]考察了烯丙基醚作为烯醚前体,在碘试剂的作用下转化成缩醛。将乙酸酯6脱乙酰基,经氢化钠作用后与烯丙基溴反应得到烯丙基醚,再用(Ph3P)3RhCl-nBuLi处理得到1-丙烯基醚11[17]。然后用NIS处理很平稳地得到碘亚乙基混合缩醛。另外,使用三氟甲磺酸二(三甲基吡啶)合碘(由I2、AgOTf和三甲基吡啶现场制备)也很有效。在I2、AgOTf和三甲基吡啶的作用下,11与氨基葡萄糖衍生物12反应以80%的收率得到混合缩醛13。接着在DTBMP存在下,由I2、AgOTf催化分子内糖苷化反应,以66%的收率得到β-甘露糖β-Man-(1(4)-GlcNAc(图4)[18]。
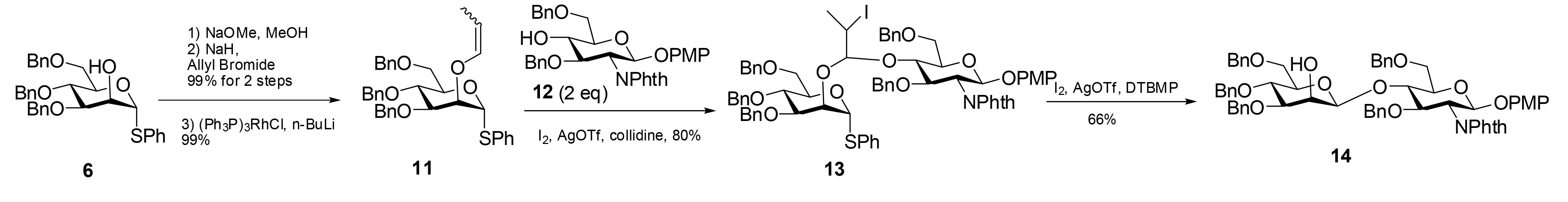
图4 通过2-碘丙基缩醛连接臂中间体的分子内糖苷配体转移
2.2 2-碘亚乙基缩醛
作为一个空间位阻最小的连接臂前体,Fairbanks等[19]考察了乙烯基醚。例如:乙烯基醚17(通过16由Ishii的铱催化的乙烯基转移作用合成[20])。然后在DTBMP存在和I2-AgOTf催化下与受体18连接得到碘亚烷基混合缩醛中间体19,接着用分子内糖苷配体转移法得到唯一的β-甘露糖苷20(图5)。
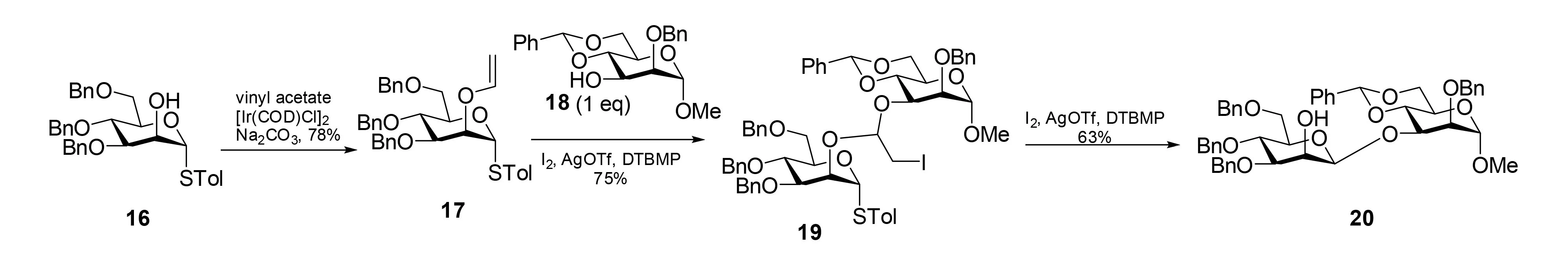
图5 通过2-碘乙烯基-连接臂中间体的分子内糖苷配体转移
2.3 2-碘-2-亚丙烯基缩醛
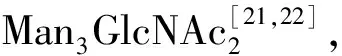
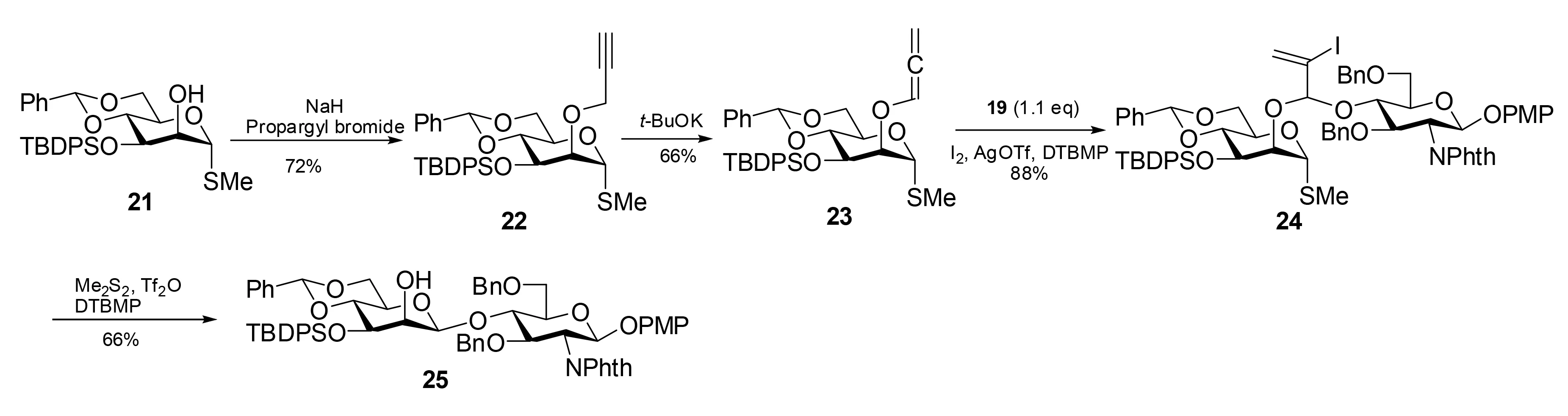
图6 通过2-碘-2-亚丙烯基连接臂中间体的分子内糖苷配体转移
3 苄叉基缩醛
3.1 对甲氧基苄叉基缩醛
在Hindsgaul和Stork报道他们早期合成β-甘露糖苷不久,Ito和Ogawa就报道了一种Hindsgaul化学的代替方法。他们用反应活性很好的对甲氧基苄基(PMB)醚代替前面所述的各种醚[23](图7)[24]。因为PMB在保护羟基中应用很广泛,在含水的二氯甲烷中他们很容易被DDQ脱出,因此,希望2-位羟基被PMB保护的甘露糖供体在无水条件下与醇反应得到混合缩醛。的确,供体-受体与DDQ混合后以非常高的收率得到混合缩醛。接下来活化异头碳位置(硫代苷或氟代苷)启动分子内糖苷配体转移,即将糖苷受体从对甲氧基混合缩醛转移到异头碳位置,得到想要的β-吡喃甘露糖苷。
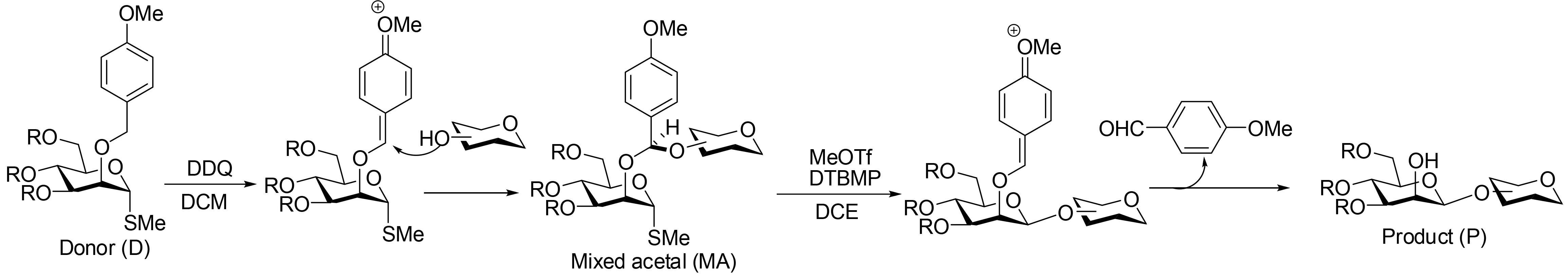
图7 通过对甲氧基苄叉基为连接臂中间体的分子内糖苷配体转移法
3.2 萘甲基缩醛
作为以PMB为基础的方法的延伸,Ito等后来将他们的研究集中到用2-萘甲基作为连接臂[25-27]。作为羟基的保护基萘甲基醚已逐渐普及到天然产物的合成[28]。与PMB醚相似。它也是在氧化条件下脱除[29,30]。因此,他们希望用与PMB相似的条件用于NAP作为连接臂的分子内糖苷配体转移法[31,32]。
事实上,2-O-NAP保护的甘露糖硫代苷供体26受体12定量地形成混合缩醛,接着在MeOTf-DTBMP催化下发生分子内糖苷配体转移,干净地得到β-甘露糖苷,在乙酰化后分离出28(图8)。像2-O-PMB保护的供体一样,混合缩醛27也是立体一致的S构型。令人欣慰的是NAP辅助的IAD法的效率要明显好于PMB辅助的反应。只要1.05当量的2-O-NAP保护的甘露糖供体26就能以90%的收率得到β-甘露糖苷28。
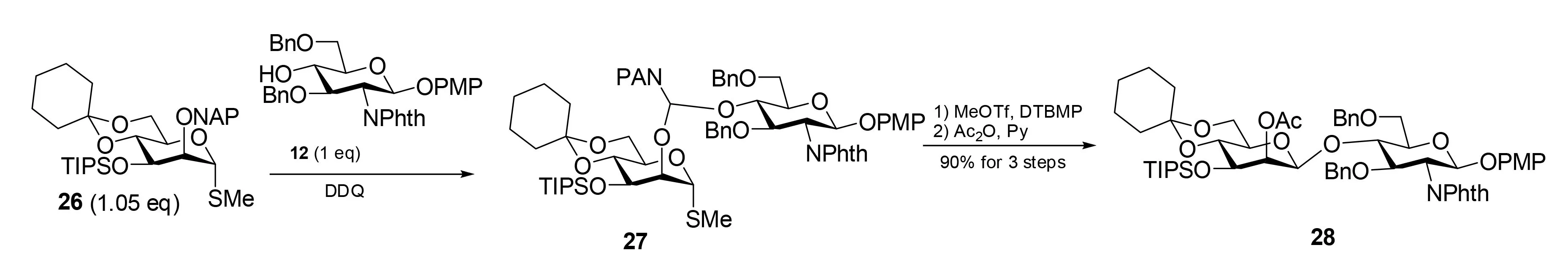
图8 萘甲基缩醛介导的分子内糖苷配体转移
4 总结与展望
通过混合缩醛(或缩酮)连接的分子内糖苷配体转移法已经在选择性合成β-甘露糖苷键中广泛应用。尽管这是一种间接方法,需要至少2步反应(即连接形成混合缩醛(或缩酮)和分子内糖苷化)才能完成反应,但是它是最有效地得到单一β-甘露糖苷键的方法,它可用于各种具有生物活性的复杂多聚糖的合成。
参考文献:
[1]TOSHIMA K, TATSUTA K. Recent progress in O-glycosylation methods and its application to natural products synthesis [J]. Chem Rev, 1993(4):1503-1531
[2]SCHMIDT R R. New methods for the synthesis of glycosides and oligosaccharides-Are there alternatives to the Koenigs-Konrr method [J]. Angew Chem Int Ed Engl, 1986(3):212-235
[3]FRASER-REID B, TATSUTA K. Thiem J Glycoscience [M]. Berlin: Springer,2001
[4]CUMPSTEY I. Intramolecular glycon delivery [J]. Carbohydr Res,2008(10):1553-1573
[5]CARMONA A T, MORENO V A J, ROBINA I. Glycosylation methods in oligosaccharides synthesis Part II [J]. Curr Org Synth, 2008(5):33-63
[6]COX L R, LEY S V. Templated Organic Synthesis [M]. New York: John Wiley & Sons,1999
[7]BOLS M, SKRYDSTRUP T. Silicon tethered reaction [J]. Chem Rev, 1995(5):1253-1277
[8]ISHIWATA A, LEE Y-J, ITO Y. Recent advances in stereoselective glycosylation through intramolecular aglycon delivery [J], Org Biomol Chem, 2010,16:3596-3608
[9]LICHTENTHALER F W. 2-Oxoglycosyl (“Ulosyl”) and 2-Oximinoglycosyl Bromides Versatile Donors for the Expedient Assembly of Oligosaccharides with β-D-Mannose, β-L-Rhamnose, N-Acetyl-β-D-mannosamine[J]. Chem Rev 2011,9:5 569-5 609
[10]BARRESI F, HINDSGAUL O. Synthesis ofβ-mannopyranosides by intramolecolar aglycon delivery [J]. J Am Chem Soc, 1991,24:9376-9377
[11]MARRA A, ESNAULT J, Veyri`eres A, et al. Isopropenyl glycosides and congeners as novel classes of glycosyl donor-theme and variations [J]. 1992,16:6354-6360
[12]JUNG M E, GERVAY J. gem-Dialkyl effect in the intramolecular Diels-Alder reaction of 2-furfuryl methyl fumarates: the reactive rotamer effect, the enthalpic basis for acceleration, and evidence for a polar transition state [J]. J Am Chem Soc, 1991(1):224-232
[13]ENNIS S C, FAIRBANKS A J. Stereoselective synthesis of alpha-glucosides and beta-mannosides: Tethering and activation with N-iodosuccinimide [J]. Synlett, 1999(9):1387-1390.
[14]ENNIS S C, FAIRBANKS A J, Slinn C A, et al. N-Iodosuccinimide-mediated intramolecular aglycon delivery [J]. Tetrahedron, 2001,19:4221-4230
[15]CUMPSTEY I, FAIRBANKS A J, Redgrave A J. Stereospecific synthesis of 1,2-cis glycosides by allyl-mediated intramolecular aglycon delivery[J]. Org Lett, 2001,15:2371-2374
[16]BOLS M. Efficient stereocontrolled glycosylation of secondary sugar hydroxyls by silicon tethered intramolecular glycosylation [J]. Tetrahedron, 1993, 49, 10049-10060
[17]BOONS G J, ISLES S. Stereospecific synthesis of 1,2-cis glycosides by allyl-mediated intramolecular aglycon delivery[J]. J Org Chem, 1996,13:4262-4271
[18]ALOUI M, CHAMBERS D J, CUMPSTEY I, et al. Stereoselective 1,2-cis glycosylation of 2-O-allyl protected thioglycosides [J]. Chem-Eur J, 2002,11:2608-2621
[19]CUMPSTEY I, CHAYAJARUS K, FAIRBANKS A J, et al. Allyl protecting group mediated intramolecular aglycon delivery: optimisation of mixed acetal formation and mechanistic investigation [J]. Tetrahedron: Asymmetry, 2004,20:3207-3221
[20]TATAI J, FUGEDI P. A new powerful glycosylation method: Activation of thioglycosides with dimethyl disulfide-triflic anhydride [J]. Org Lett, 2007,22:4647-4650
[21]CHAYAJARUS K, ChAMBERS D J, CHUGHTAI M J, et al. Stereospecific synthesis of 1,2-cis glycosides by vinyl-mediated IAD [J]. Org Lett, 2004,21:3797-3800
[22]OKIMOTO Y, SAKAGUCHI S, ISHII Y. Development of a highly efficient catalytic method for synthesis of vinyl ethers [J]. J Am Chem Soc, 2002(8):1590-1591
[23]ATTOLINO E, FAIRBANKS A J. β-Mannosylation of N-acetyl glucosamine by propargyl mediated intramolecular aglycon delivery (IAD)[J]. Tetrahedron Lett, 2007,17:3061-3064
[24]HIRAMA M, OISHI T, UEHARA H, et al. Total synthesis of ciguatoxin CTX3C [J]. Science, 2001(8):1904-1907
[25]INOUE M, UEHARA H, MARUYAMA M, et al. Practical total synthesis of ciguatoxin CTX3C by improved protective group strategy [J]. Org Lett, 2002,25:4551-4554
[26]INOUE M, MIYAZAKI K, UEHARA H, et al. First- and second-generation total synthesis of ciguatoxin CTX3C [J]. Proc Natl Acad Sci USA,2004,33:12013-12018
[27]GAUNT M J, YU J, SPENCER J B. Rational design of benzyl-type protecting groups allows sequential deprotection of hydroxyl groups by catalytic hydrogenolysis [J]. J Org Chem, 1998,13:4172-4173
[28]XIA J, ABBAS S A, LOCKE R D, et al. Total synthesis of a sialylated and sulfated oligosaccharide from O-linked glycoproteins [J]. Tetrahedron Lett, 2000(2):169-173
[29]CRICK D, VINOGRADOVA O. Facile oxidative cleavage of 4-O-benzyl ethers with dichlorodicyanoquinone in rhamno- and mannopyranosides [J]. J Org Chem, 2007,17:3581-3584
[30]BOECKMAN R K, CLARK T J, SHOOK B. The development of a convergent and efficient enantioselective synthesis of the bengamides via a common polyol intermediate [J]. Helv Chim Acta, 2002,12:4532-4560
[31]KULKARNI S S, LIU Y H, HUNG S C. Neighboring group participation of 9-anthracenylmethyl group in glycosylation: Preparation of unusual C-glycosides [J]. J Org Chem, 2005(7):2808-2811
猜你喜欢
现代食品科技(2023年12期)2024-01-09 01:22:38
中国药学药品知识仓库(2022年10期)2022-05-29 02:59:32
大学化学(2022年1期)2022-02-28 12:16:50
汕头大学学报(自然科学版)(2020年4期)2020-12-14 07:04:58
汕头大学学报(自然科学版)(2020年4期)2020-12-14 07:04:58
合成树脂及塑料(2020年1期)2020-03-27 14:14:02
化工管理(2017年12期)2017-03-04 00:07:45
分析科学学报(2015年3期)2015-10-18 02:25:40
四川师范大学学报(自然科学版)(2014年4期)2014-10-09 01:19:56
无机化学学报(2014年12期)2014-02-28 17:34:01
