
液相色谱-串联质谱法检测动物源性水产品中7种微囊藻毒素
2014-08-07 03:11:50杨振宇
质谱学报 2014年5期
杨振宇,周 瑶
(上海出入境检验检疫局,上海 200135)
近年来,水体富营养化现象出现在世界许多大水系中。水体出现富营养化现象时,藻类大量繁殖,产生的一些溶于水的代谢物(如藻毒素)不仅可使水体生物死亡,甚至可以通过饮用水和水产品富集危害人类健康。在水华藻类中,毒性最强、污染范围最广的是蓝藻(blue-green algae)。蓝藻广泛分布于江河、湖沼、海洋等水体中,大多数蓝藻在代谢过程中能产生各种毒素,其中,微囊藻毒素(microcystin,MC)是蓝藻释放的一类具有强烈致癌作用的肝毒素和神经毒素。
MC是一种环肽肝毒素,其结构为环(D-丙氨酸-L-R1-赤-β-甲基-D-异天冬氨酸-L-R2-Adda-D-异谷氨酸-N-甲基脱氢丙氨酸)。其中,N-甲基脱氢丙氨酸(Mdha)是一种特殊的氨基酸,含有α、β不饱和双键;Adda结构为3-氨基-9-甲氧基-2,6,8-三甲基-10-苯基-4,6-二烯酸,是MC生物毒性表达所必需的;R1、R2为2种可变L-氨基酸,由于R1、R2位置上的氨基酸不同,环状结构不同位置侧链的不同,以及侧链上甲基化/去甲基化产生的差异,可以形成多种不同结构。目前已从不同微囊藻菌株中分离鉴定了近80种MC结构。本实验将检测的7种MC结构中R1与R2的氨基酸种类列于表1。
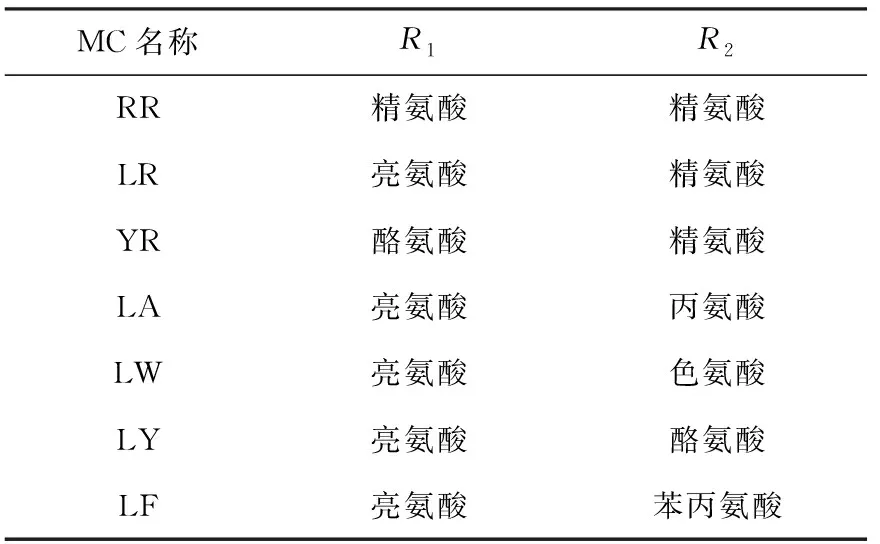
表1 7种MC结构中R1与R2的氨基酸种类
MC具有水溶性和耐热性,易溶于甲醇,可溶于水。由于环状结构和间隔双键,其结构具有相当的稳定性[1-2]。肝脏是MC主要的靶器官,所以肝毒性是MC的主要毒性,在低浓度时就有专有肝毒性和癌诱发活性。同时,MC也表现出胚胎发育、免疫、遗传等方面的毒性[1-3]。MC不仅能直接导致一些水生生物(如鱼类、贝类及蚌类)患病及死亡,而且一些野生动物及家畜饮用了含有藻毒素的水后,会引起中毒甚至死亡,同时还可能通过食物链的生物富集危害人类的健康。
MC的检测方法较多[4-6],可分为液相检测方法[7-8]、生物学方法[9]和免疫检测法[10-11]等。液相色谱-串联质谱由于定性准确、定量灵敏,是微量毒素分析最有效的检测技术[11-18]。本实验拟采用固相萃取和基质分散固相萃取两种前处理方法,用液相色谱-串联质谱法检测水产品中7种MC。
1 实验部分
1.1 主要仪器与装置
TSQ Quantum Ultra液相色谱-串联质谱仪:美国Thermo-Fisher公司产品,配有Accela液相色谱仪;Allegla X-22R高速离心机:美国Beckman Coulter公司产品;PT2100均质仪:Poly TRO公司产品;QGC-12T氮吹仪:上海泉岛公司产品;890/H超声仪:德国Elma公司产品;KMC-1 300 V涡旋振荡器: Vision公司产品;固相萃取装置:美国Supelco公司产品;Waters Oasis HLB固相萃取小柱:美国Waters公司产品;DSPE PSA/C18 MSPD基质分散固相萃取管:德国CNW公司产品;水相针式滤膜(聚醚砜材质,13 mm×0.22 μm):德国CNW公司产品。
1.2 主要材料和试剂
水:满足GB/T 6682规定的一级水要求;甲醇:色谱纯;甲酸:色谱纯,含量≥98%;正己烷:分析纯。
MC标准品(MC-RR、LR、YR、LA、LW、LF、LY微囊藻毒素固体标准): 瑞士Alexis公司产品。MC-RR、MC-LR为0.1 mg,其它均为0.025 mg,使用前用甲醇稀释,配成7种MC混合标准溶液,20%甲醇定容。
1.3 实验条件
1.3.1色谱条件 色谱柱:Hypersil GOLD C18 (50 mm×2.1 mm×1.9 μm);柱温35 ℃;进样量10 μL;流动相:0.1%甲酸水溶液(A)和甲醇(B);梯度洗脱程序:0~3 min、20%B,3~5 min、20%~95%B,5~5.1 min、95%~20%B,5.1~7 min、20%B。
1.3.2质谱条件 电喷雾正离子模式,多反应监测,喷雾电压3.5 kV,喷雾温度300 ℃,鞘气气压30,辅助气压10,毛细传输管温度270 ℃。母离子、子离子和相关参数列于表2。
1.4 前处理方法
1.4.1样品匀浆 准确称取2 g淡水产品的食用部分于50 mL离心管中,加入5 mL 80%甲醇水溶液,匀质器上以20 000 r/min混合1 min,使样品均质成匀浆,然后超声15 min,以4 000 r/min离心5 min。
1.4.2固相萃取法(SPE) 将1.4.1离心后的上清液转移至试管中,70 ℃氮吹至约0.5 mL,加水至约10 mL,待SPE柱净化。HLB固相萃取小柱在使用前应分别用5 mL甲醇和水以1~2 mL/min流速淋洗活化,活化后的小柱应尽快使用。将10 mL样液转移到HLB小柱上,控制上样流速为1~2 mL/min;用5 mL 10%甲醇以1~2 mL/min流速淋洗,抽干;用5 mL 80%甲醇溶液以1 mL/min流速洗脱;收集洗脱液,并于70 ℃下氮吹至近干;最后用20%甲醇水溶液定容至1 mL,过0.22 μm滤膜,供液相色谱-质谱仪测定。
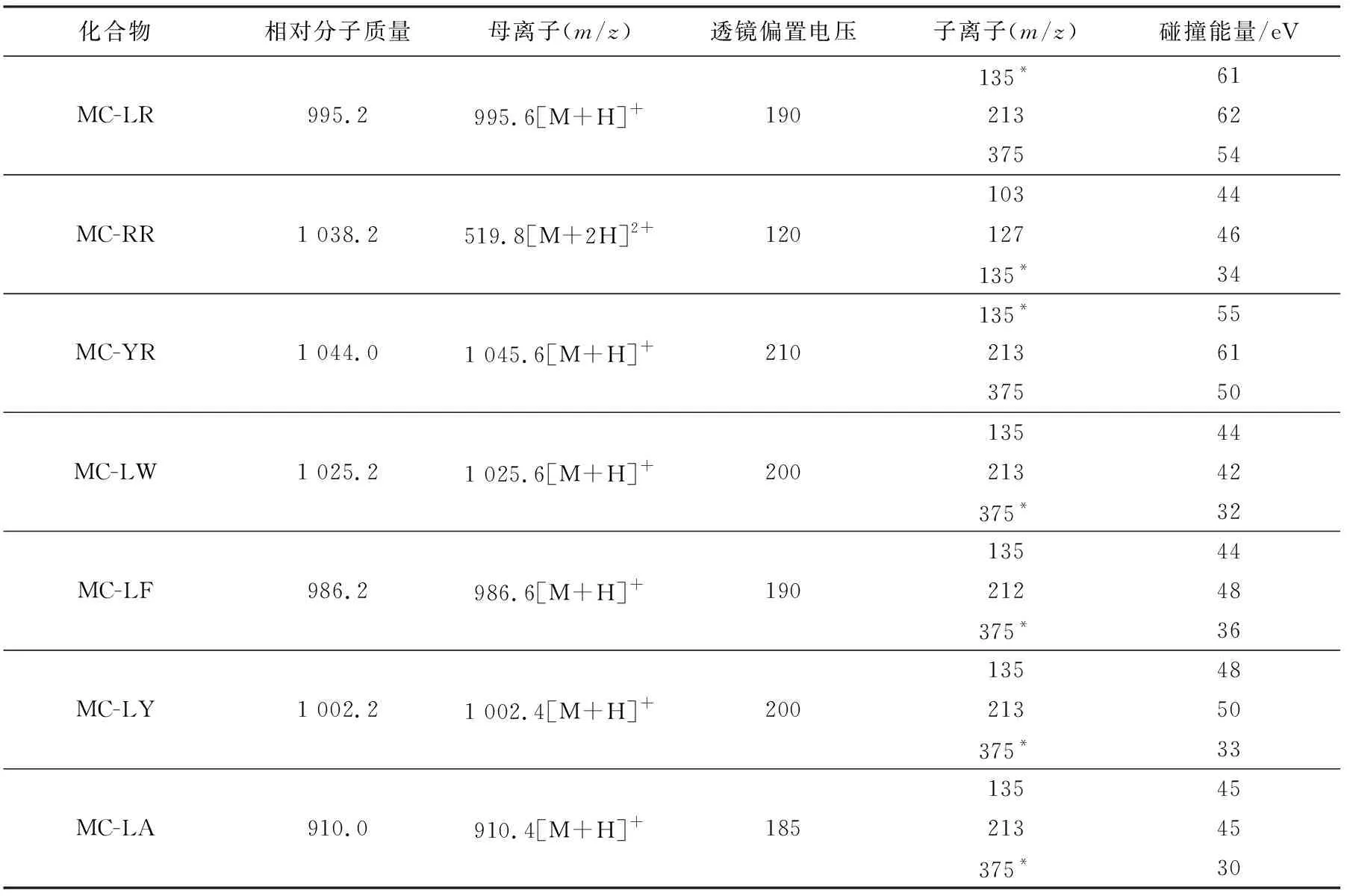
表2 7种MC分析物的母离子、子离子和相关参数
注:*为定量离子
1.4.3基质分散固相萃取法(MSPD) 将1.4.1离心后的上清液转移到DSPE PSA/C18 MSPD基质分散固相萃取管中,振摇2 min,以4 000 r/min离心5 min,取全部上清液至10 mL比色管中,用水定容至5 mL。混匀后,取1 mL溶液过0.22 μm滤膜,供液相色谱-质谱仪测定。
2 结果和讨论
2.1 质谱条件的优化
配制10 mg/L的MC标准溶液,分别对每个MC标准溶液进行母离子和子离子的参数优化。
由于ESI离子源是一种软电离方式,大部分目标化合物会形成1价离子,也可能形成2价离子、多价离子或者其他加合离子。由表2可以看出,大部分MC主要形成加氢的1价正离子,但MC-RR的离子形成与其他离子不同,它产生2价离子的产率要比形成其他离子的高,所以选用m/z519.8[M+2H]2+作为MC-RR母离子。
子离子以m/z135、213、375为主,这3种子离子都是MC的特征子离子,具体分子式和结构见表3和图1。m/z135是MC的共有基团3-氨基-9-甲氧基-2,6,8-三甲基-10-苯基-4,6-二烯酸(Adda)的甲氧键断裂形成的特征碎片,它在MC-LR、MC-RR、MC-YR子离子质谱图上的丰度最大,其他MC的子离子也含有此离子。另外,m/z213、375也是大部分MC共有结构的特征碎片,所以本实验所检测的7种MC的子离子也都有这2种质量数的碎片。MC-RR、LR、YR将m/z135作为定量离子,其他4种MC用m/z375作为定量离子。
为了得到较高的离子化效率,在使用ESI离子源时,需加入一些离子化增强试剂。本实验采用甲酸作为离子化试剂,并对流动相中的甲酸含量进行优化实验,不同甲酸浓度对灵敏度的影响示于图2。
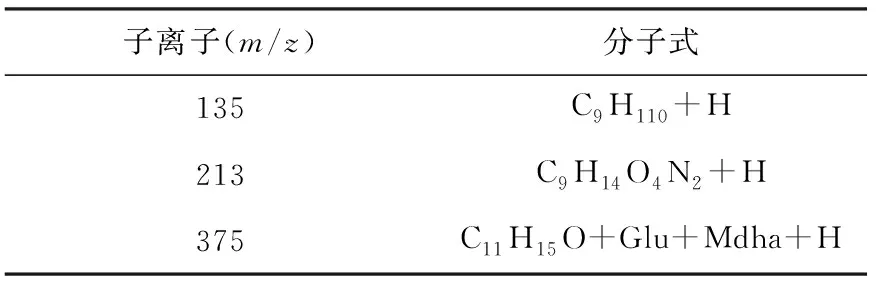
表3 子离子的分子式
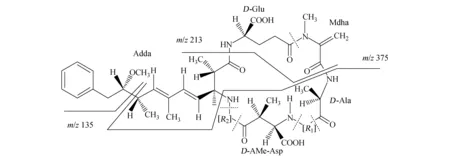
图1 3种子离子可能的碎裂方式Fig.1 The possible fragmentation of daughter ions
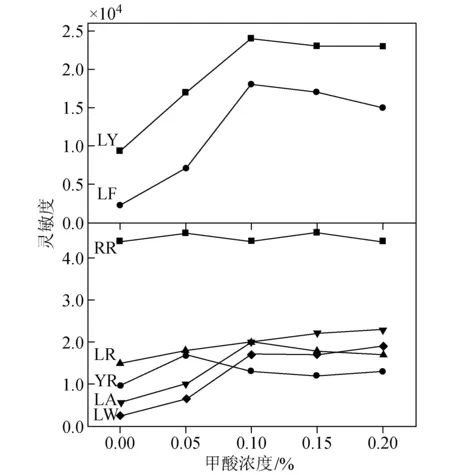
图2 不同甲酸浓度对灵敏度的影响 Fig.2 Effect of different concentrations of formic acid on sensitivity
从图2可以看到,加入甲酸后,MC-LA、LY、LF、LW的灵敏度大大增加,但在0.1%和0.2%甲酸含量水平,灵敏度变化不大。所以采用在流动相中添加0.1%甲酸,优化后的总离子流图和定量离子色谱图示于图3。
2.2 前处理方法的选择
根据文献[2]和MC本身的特性,SPE柱一般用C18小柱或者HLB小柱净化和富集样品。本实验对HLB小柱的淋洗条件做了回收实验,步骤如下:在活化的HLB小柱上,上样10 mL含7种MC的水样,其中每种MC各20 ng,然后分别用5 mL 100%水、10%、20%、30%、40%、50%、60%、70%、80%、90%甲醇水溶液、100%甲醇进行洗脱,分别收集洗脱液,上机测量MC含量,并计算洗脱液中7种MC含量。结果发现:RR在50%~80%甲醇水淋洗段被淋洗下来;YR、LR、LA、LF、LW、LY在20%~80%甲醇水淋洗段被淋洗下来。所以本实验使用10%甲醇水溶液淋洗,80%甲醇水溶液洗脱。
目前,基质分散固相萃取方法(matrix solid phase dispersion,MSPD)已被用于多种残留分析的前处理[19-20],在MC检测中,MSPD也有一些应用[21-22]。
本实验采用CNW公司的DSPE PSA/C18 MSPD基质分散固相萃取管。这是一根容积为15 mL的离心管,里面灌装了150 mg PSA、150 mg C18、900 mg无水硫酸镁等固体粉末。当样品的甲醇水提取液倒入MSPD管,C18会吸附其中的非极性物质,PSA(乙二胺-N-丙基)会吸附脂肪酸、有机酸、极性色素、糖类等物质,硫酸镁则会吸附水,同时吸附水溶性杂质,而MC则会留在甲醇溶液中,从而达到净化的目的。需要指出的是,该净化效果不如过柱法好,检出限差,回收率相对较低,但该过程的检测时间短、操作方便、检出限符合要求,是快速简便的方法。
2.3 方法指标
根据不同前处理方法导致的不同检出限,设置了不同的添加水平,对水产品中MC进行回收率和精密度实验。SPE法的添加水平为0.5、1、2 μg/kg;MSPD法的添加水平为2.5、5、10 μg/kg,对应上机溶液中的MC含量水平为1、2、4 μg/L。选取了均为阴性样品的笋壳鱼肉和蛳螺肉作为添加样品基质,平行测量6次,结果列于表4。
由表4可以看到出:SPE法的回收率为75%~106%,相对标准偏差小于20%;MSPD法的回收率为73%~93%,相对标准偏差小于19%。
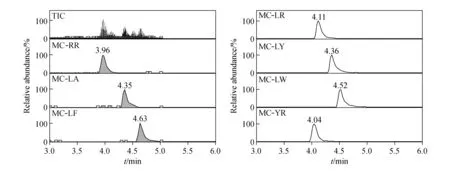
图3 7种MC的色谱图Fig.3 Chromatograms of 7 MCs
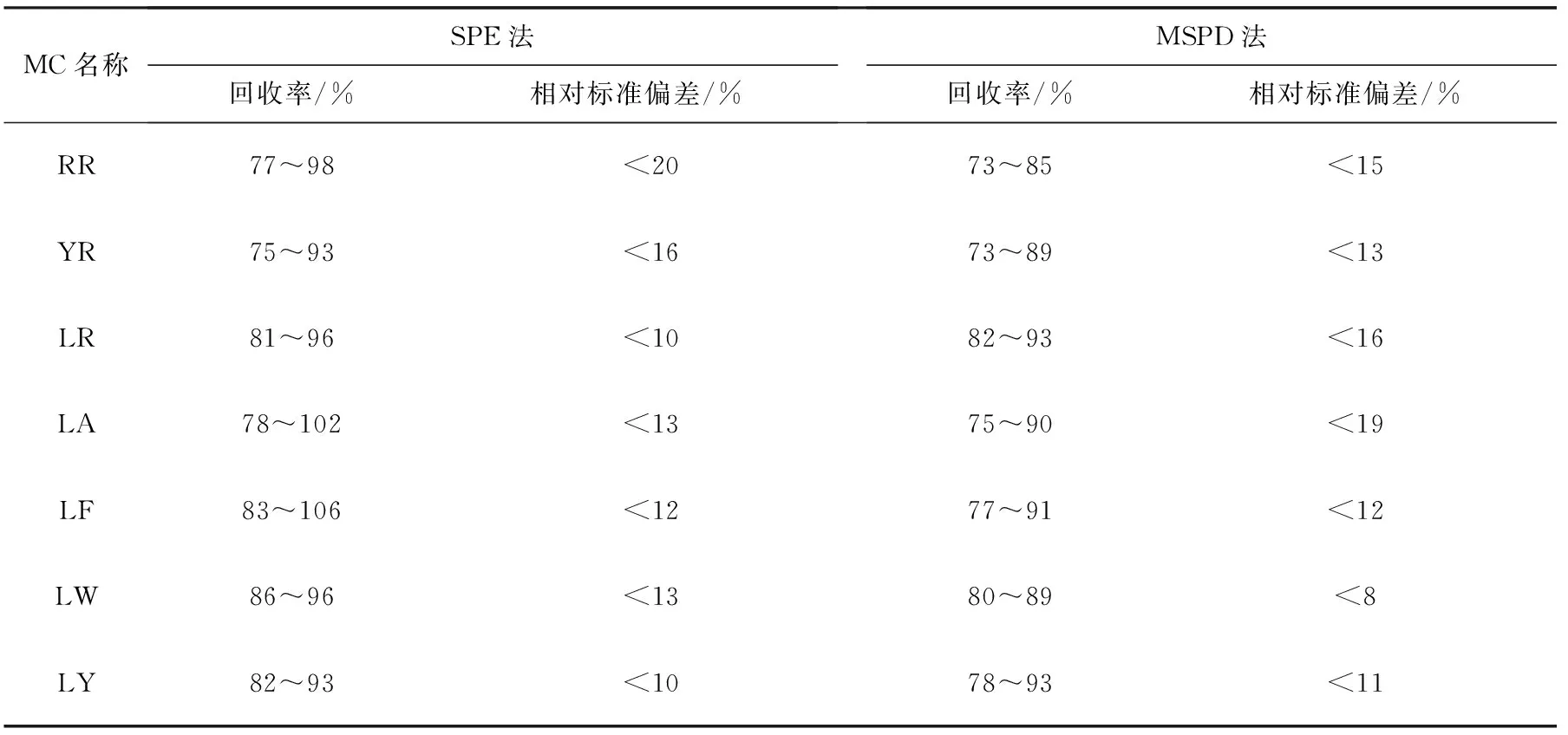
MC名称SPE法回收率/%相对标准偏差/%MSPD法回收率/%相对标准偏差/%RR77~98<2073~85<15YR75~93<1673~89<13LR81~96<1082~93<16LA78~102<1375~90<19LF83~106<1277~91<12LW86~96<1380~89<8LY82~93<1078~93<11
3 结论
建立了液相色谱-串联质谱测定动物源性水产品中7种MC的方法,7种MC在0.5~20 μg/L范围内线性良好。根据动物源性水产品样品的情况,建立了SPE和MSPE两种前处理方法。SPE法操作较繁琐,测量过程较长,但检测限较低,为0.5 μg/kg,添加回收率为75%~106%,相对标准偏差小于20%;而MSPD法操作简单,但检测限则不如SPE法,为2.5 μg/kg,添加回收率为73%~93%,相对标准偏差小于19%。因此,可以根据实际要求,选取不同的前处理方法进行检测。
参考文献:
[1] 李效宇. 微囊藻毒素及其毒理学研究[M]. 北京:科学出版社,2007.
[2] 谢 平. 水生动物体内的微囊藻毒素及其对人类健康的潜在威胁[M]. 北京:科学出版社,2006.
[3] 张敬平, 肖付刚, 赵晓联. 微囊藻毒素分析检测技术[M]. 北京:化学工业出版社,2010.
[4] MORENO I M, MOLINA R, JOS A, et al. Determination of microcystins in fish by solvent extraction and liquid chromatography[J]. J Chromatogr A, 2005,1 080(2):199-203.
[5] 虞锐鹏, 陶冠军, 贡小清,等. 高效液相色谱-质谱联用方法测定背角无齿蚌体内微囊藻毒素[J]. 分析测试学报, 2007,26(5):671-674.
YU Ruipeng, TAO Guanjun, GONG Xiaoqing, et al. Determination of microcystins in anodonta woodiana by high performance liquid chromatography combined with electrospray ionization mass spectrometry[J]. Journal of Instrumental Analysis, 2007, 26(5):671-674(in Chinese).
[6] MEKEBRI A, BLONDINA G J, CRANE D B. Me-thod validation of microcystins in water and tissue by enhanced liquid chromatography tandem mass spectrometry[J]. J Chromatogr A, 2009,1 216(15):3 147-3 155.
[7] 刘晓蕙. 多聚酶链反应(PCR)技术监测水中微囊藻毒素的方法及进展[J]. 河南预防医学杂志, 2004,15(6):365-367.
LIU Xiaohui. Method and progress of determinate the microcystins in water by PCR technical[J]. Henan J Prev Med, 2004,15(6):365-367(in Chinese).
[8] 闫建秀, 虞锐鹏, 汤 坚, 等. 固相萃取-反相高效液相色谱法检测太湖蛳螺中微囊藻毒素[J]. 食品与机械,2008, 24(5):92-95.
YAN Jianxiu, YU Ruipeng, TANG Jian, et al. Determination of microcystins in the snail genus eriocheir from Taihu using RP-HPLC with an efficient solid phase extraction[J]. Food & Machinery,2008, 24(5):92-95(in Chinese).
[9] 王 蕾,李小艳,薛文通,等. 微囊藻毒素检测方法研究进展[J]. 食品科学,2005,26(增刊):135-138.
WANG Lei, LI Xiaoyan, XUE Wentong,et al. Review on research progress of microcystins detection[J]. Food Science, 2005, 26(Suppl):135-138(in Chinese).
[10] 赵晓联,孙秀兰,汤 坚. 藻毒素的危害及分析方法进展[J]. 食品科学,2005, 26(3):257-261.
ZHAO Xiaolian, SUN Xiulan, TANG Jian. Development of assay methods and hazards of microcystins[J]. Food Science, 2005, 26(3):257-261(in Chinese).
[11] BRUNO M, FIORI M, MATTEI D, et al. EL-ISA and LC-MS/MS methods for determining cyanobacterial toxins in blue-green algae food supplements[J]. Natural Product Research, 2006, 9(20): 827- 834.
[12] DAI M, XIE P, CHEN J, et al. Quantitative determination of microcystins in rat plasma by LC-ESI tandem MS[J]. Chromatographia, 2008, 68(9/10): 811-815.
[13] 虞锐鹏, 陶冠军, 杨 健, 等. 超高效液相色谱-四极杆-飞行时间质谱法快速测定水产品中微囊藻毒素和节球藻毒素[J]. 分析试验室,2012,31(1):80-83.
YU Ruipeng, TAO Guanjun, YANG Jian, et al. Determination of microcystins and nodularins in aquatic products by ultra performance liquid chromatography-quadrupole-time of flight mass spectrometry[J]. Chinese Journal of Analysis Laboratory, 2012, 31(1): 80-83(in Chinese).
[14] 明俊超, 姜海洲, 袁新华. 微囊藻毒素对鱼类的毒性效应及其作用机理研究进展[J]. 中国农学通报, 2012,28(35):69-74.
Ming Junchao, Jiang Haizhou, Yuan Xinhua. A review of microcystin of toxic effects and mechanisms in exposed fish[J]. Chinese Agricultural Science Bulletin, 2012,28(35):69-74(in Chinese).
[15] WELLER M G. Immunoassays and biosensors for the detection of cyanobacterial toxins in water[J]. Sensors, 2013, (13): 15 085-15 112.
[16] XU W, CHEN Q, ZHANG T, et al. Development and application of ultra performance liquid chromatography-electrospray ionization tandem triple quadrupole mass spectrometry for determination of seven microcystins in water samples[J]. Analytica Chimica Acta, 2008, 626(1):28-36.
[17] DAI M, XIE P, LIANG G D, et al. Simultaneous determination of microcystin-LR and its glutathione conjugate in fish tissues by liquid chromatography-tandem mass spectrometry[J]. Journal of Chromatography B, 2008, 862(1/2):43-50.
[18] 钮伟民,肖付刚,戴维杰, 等. 液相色谱-串联质谱法同时检测水中5种微囊藻毒素[J]. 安徽农业科学, 2008,36(29):12 554-12 556.
NIU Weimin, XIAO Fugang, DAI Weijie, et al. Simultaneous deterndnation of nve microcystins in water by mgh perfornmnce liquid chromatography-tandem mass spectrometry [J]. Journal of Anhui Agri Sci, 2008,36(29):12 554-12 556(in Chinese).
[19] 李 尧, 张雪梅, 党献民, 等. 基质分散固相萃取净化液相色谱检测谷物中赭曲霉毒素A[J]. 粮食与饲料, 2012,(10):57-60.
LI Yao, ZHANG Xuemei, DANG Xianmin, et al. Determination of ochratoxin A in cereal by high performance liquid chromatography with matrix solid-phase dispersion cleaning [J]. Cereal & Feed industry, 2012,(10):57-60 (in Chinese).
[20] 乔凤霞, 孙汉文, 刘广宇, 等. 基质固相分散-牛奶、蜂蜜中喹诺酮的多残留分析[J]. 河北大学学报:自然科学版,2008, 28(6):620-624.
QIAO Fengxia, SUN Hanwen, LIU Guangyu, et al. Matrix solid-phase dispersion for multi-residues analysis of quinolones in milk and honey [J]. Journal of Hebei University(Natural Science Edition), 2008, 28(6):620-624(in Chinese).
[21] CAMEN A, MORENO I M, RUIZ M J, et al. Determination of microcystins in natural blooms and cyanobacterial strain cultures by matrix solid-phase dispersion and liquid chromatography-mass spectrometry[J]. Anal Bioanal Chem, 2004, 380(3):537-544.
[22] RUIZ M J, CAMEN A M, MORENO I M, et al.
Determination of microcystins in biological samples by matrix solid-phase dispersion and liquid chromatography-mass spectrometry[J]. Journal of Chromatography A, 2005, 1 073(1/2):257-262.
猜你喜欢
少年文艺(2022年8期)2022-07-08 10:02:47
中国蜂业(2018年4期)2018-05-09 06:25:08
中国经济周刊(2017年6期)2017-03-21 00:59:27
读写算·高年级(2016年3期)2016-05-30 01:53:46
当代化工研究(2016年6期)2016-03-20 16:21:46
中国医疗美容(2015年1期)2015-07-12 10:06:18
西北园艺(果树)(2015年1期)2015-02-21 16:44:50
癌变·畸变·突变(2014年2期)2014-03-01 04:39:43
无机化学学报(2014年3期)2014-02-28 17:30:58
河南科技(2014年12期)2014-02-27 14:10:32
