
GABAA受体研究现状
2014-08-06 07:18袁莲芳贾宏阁刘利宁李毓新师建国
现代临床医学 2014年2期
袁莲芳,陈 果,贾宏阁,高 影,刘利宁,李毓新,师建国
(1.西安医学院第二附属医院,陕西 西安 710038;2.第四军医大学西京医院,陕西 西安 710032)
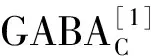
1 GABAA受体的分子结构
GABAA受体是由GABA识别点、苯二氮(BDZ)识别点和Cl-门控通道3部分组成的大分子蛋白复合体。目前的研究结果证明GABAA受体是由镶嵌于神经细胞质膜双脂层中的5个亚基(来自8个亚基族)组成的五边形异质性多肽类寡聚体,其中心部位形成一个直径为0.5 nm的GABA门控Cl-通道[2],这5个亚基由2个α亚基(α1~6)、2个β亚基(β1~3)或θ亚基和1个γ亚基(γ1~3)/δ或ε亚基组成[3]。
2 GABAA受体的配体
3 GABAA受体的活性部位
药理学分析和定点突变研究证实GABAA受体上有一些氨基酸残基在GABA和BDZ类药物与受体的结合过程中起重要作用。文献报道,α亚基上的His101 和Gly200与γ亚基上的he77、Met130和Thr142是大鼠GABAA受体BDZ结合位点形成的关键氨基酸,α1亚基上Tyr159、yr161、Thr162、Thr206、Thr209、Val211影响配体与BDZ位点结合的亲和力,β亚基上的Tyr157、Thr160、Thr202与Tyr205和α1亚基上的Phe64是GABA与GABAA受体结合所必需的氨基酸。此外,实验结果表明受体上的一些氨基酸残基在抑制剂巴比妥类以及神经活性类固醇类药物等与GABAA受体结合时发挥重要作用[6]。
4 GABAA受体的亚基
通过cDNA克隆技术已测定出在哺乳动物大脑内有23个肽GABAA受体亚基的氨基酸基因序列[7],依氨基酸序列相似程度,将亚基系列分为8个亚基族,分别命名为α1~6、β1~6、γ1~3、δ1、ε1、π1、ρ1~3、θ1,由400~550 个氨基酸残基组成,其中包括约220个氨基酸残基组成的胞外N端疏水域,含有2个(α1亚基)、3个(α2亚基、β亚基)和4个(α3亚基)N糖基化部位,都具有4个疏水的跨膜顺序(TM1~4),跨膜顺序彼此之间通过亲水性氨基酸相连接,TM1共同序列中心有一保守的脯氨酸存在,在受体通道形成、门控和巴比妥类作用等方面发挥重要作用,并且脯氨酸在疏水的TM1的p结构中形成一个回折,影响邻近TM2的螺旋,TM2亲水性氨基酸残基形成Cl-通道,能选择性通过带负电荷的Cl-。受体所有亚基胞外区均有一个由15个氨基酸残基形成的β环结构。γ2L亚基氨基酸序列中含有一个被蛋白激酶C磷酸化的氨基酸位点和一个被Ca2+/钙黏蛋白依赖的蛋白激酶Ⅱ的氨基酸位点[8]。哺乳动物大脑中存在的天然GABAA由α、β和γ亚基组成(α3、β2、γ2)[9-10],至少有12~24 个亚型具有重要的生理功能[11-12]。大脑中存在的受体亚型可能是由1个、2个、3个或5个不同亚基所组成的天然受体。不同的GABAA受体亚型分别表达在大脑特定的区域,多数神经元仅表达一个主要的亚型,即特定神经元表达GABAA受体含有特定的亚基组合。体外重组GABAA受体表达研究表明,亚基的组成决定受体的药理学和电生理学性质,大量的受体亚型存在很有可能建立起新的带有选择性疗效的作用靶。GABAA受体亚型的结构和药理学研究可望在大脑不同区域为选择性调节GABAA受体亚型开辟新的途径。
5 GABAA受体亚基基因
A受体基因主要由5q34~35区域α1、γ2基因簇和15q11~13区域α5、β3、γ3基因簇共同构成。GABAA受体α1基因定位于5q34~35区域。γ2定位于5q31.1~33.1区域,与α1邻近。α5、β3、γ3基因定位于15q11.2~12区域。β是GABAA受体的主要组成部分,γ3是GABAA受体中最重要的辅助结构。
5.1 α1γ2基因簇的研究 GABAA受体α1基因由Buckle等定位在5q34~35区域。α1活化主要依赖于细胞的静息电位。GABAA受体γ2基因由Warrington在1992年首次定位在5号染色体5q31.1~33.1区域。该基因参与编码GABAA受体亚型的复合配体门通道。
5.2 α5、β3、γ3基因簇的研究 GABAA受体α5基因被定位在15q11.2~12区域。目前对α5基因的认识还不多,主要是通过对Angelman和Prader-Willi综合征的研究得知二者都存在发育迟滞、严重的癫痫发作等临床表现。Feuch、Robinson和我国学者吕建军等均经过研究得出结论是GABAA的α5,β3基因可能是儿童癫痫小发作(childhood absence epilepsy,CAE)的易感基因或与之存在连锁不平衡。
6 GABAA受体亚基的变异
Baulac等2001年报道在一个以高热惊厥伴全面性癫痫为表现型的家系中发现γ2基因lys289的突变。澳大利亚的Wallace等2001年对一个CAE伴FS的家系研究发现γ2基因内的arg43发生了突变。Kananura等也报道发现CAE和高热惊厥与GABAA受体γ2基因错义突变产生的无功能性的A受体γ亚单位相关联。Baulac等通过对法国全身性癫痫家族进行研究,发现GABAA受体γ2亚基GABRG2的第8外显子的K289M发生了变异,该变异与全身型癫痫伴热性惊厥( generalized epilepsy with febrile seizures plus,GEFS+)的表型密切相关。他们的研究显示这个变异不同的神经元网络有年龄相关性。Harkin等在GEFS+家族中发现了GABRG2的Q351基因变异,这个变异在Q351引入了一个过早的终止密码子,位于M3、M4之间的细胞内环路。卵母细胞纯合子表达下,GABA敏感性消失,提示有非功能的GABAA受体。利用绿荧光蛋白标记γ亚基,他们发现受体尽管已经组装,但没有显示出外在表达,而是滞留在内质网。这就说明此变异可能导致缩减功能性GABAA受体复合物的表面表达,最终导致GABA能抑制降低、兴奋性增高。Kananura等在CAE和热性惊厥家族中发现GABRG2剪切位点变异。Chou等利用单核苷酸多态性方法研究热性癫痫(febrile seizures,FSs)患者GABRG2基因型的分布。他们用PCR法辨别染色体5q33的GABRG2基因C/T以及A/G多态性,得出GABRG2基因可能是FSs的一个易感因素的结论。Svend等通过突变反应将α亚基121位异亮氨酸突变为缬氨酸后大大降低了GABAA受体对兴奋剂的敏感性。通过α亚基的点突变可使该位点对地西泮不敏感。Cossette等报道过GABAA受体α1亚基(A322D)第三跨膜区域M3的高保守丙胺酸残基变异,此变异与常染色体显性青少年肌阵挛性癫痫有关。
7 GABAA受体在中枢神经系统的区域分布
广泛分布于整个神经系统,以小脑最高,海马其次,延髓和脊髓最低[13]。原位杂交研究表明,脑组织中各种GABAA受体mRNA具有不同但又交叉的区域分布。离体和在体显示的受体异质中,α1β2γ2受体亚型的数量最多(占43%),富含于海马和大脑皮质中。其次为α2β2γ2和α3β2γ2,约为35%,分布于海马、纹状体及大脑皮质[14]。
8 GABAA受体的测定方法
目前测定GABAA受体的方法主要为放射性配基结合受体法,测出GABAA受体的最大结合容量和平衡解离常数值。另有免疫印迹和免疫检测。用RT-PCR测定GABAA受体亚单位的基因表达也受到重视[15]。
异步改造式的SPOC设计方式是通过改造现有MOOC课程或精品课程等资源,开展SPOC教学。现有的国家精品课程或世界一流的MOOC课程在师资力量和教学质量上有着无法比拟的优势,但是这些优秀资源却很少能让普通高校的在校生直接受益。异步改造式教师根据本校课程教学的目标与需求,改造精品课程或者MOOC课程的总体框架结构、测试评价方式,以及部分的教学资源等,使得优质的教学资源融合进高校课程教学,提高普通高校课程的教学质量。
9 GABAA受体的神经药理学研究
关于GABAA受体药理学研究多集中于BDZ识别位点。研究表明,改变受体亚基组分的一个氨基酸片段,会使重组受体药理学性质得到修饰。GABAA受体是一种多功能药物作用靶标,它可以介导抗焦虑药、镇静剂、抗惊厥药、肌肉松弛和失忆作用,此介导作用通常是非选择性的,即这些药物将作用于GABAA受体家族的几种亚型。
9.1 抗焦虑作用 近年来,不少制药公司尝试着研制无镇静组分和依赖效应的新型抗焦虑药物。分子研究、动物研究和临床研究的证据表明GABAA受体复合物在调节焦虑症上起着主要作用,对抗焦虑作用的实验如明暗选择试验和增强迷宫试验[16]证明BDZ抗焦虑作用是通过能表达含α2受体的神经元群增强其GABA传导进行选择性介导的,而这些α2受体仅占所有BDZ敏感GABA受体的15%。鉴于未来抗焦虑药物将只与15%的GABAA受体结合,那将极有希望消除长期困扰BDZ药物的主要副作用,从而研制出真正意义上高效、低副作用的新型抗焦虑药物。在α3[H126R]突变小鼠与野生型小鼠的行为去抑制实验中证明含α3受体并不参与BDZ抗焦虑作用,因此,含α2而不含α3受体可作为未来选择性抗焦虑药物的一个高特异性靶标,由此得出,BDZ药物的抗焦虑效应是由边缘系统中大量表达的α2受体而非网状激活系统中占主导的α3受体介导的。
9.2 镇静作用 镇静作用是许多BDZ位点配体的主要特性。与BDZ的镇静效应相似,安眠药唑吡坦在体内也是通过α1受体介导的。
9.3 肌肉松弛作用 BDZ的肌肉松弛作用主要是由含α2、α3或α5受体介导的,因为在α1[H101R]突变小鼠肌肉紧张实验中BDZ诱发产生了变化,Bohlhalter等进一步假设含α2受体是BDZ肌肉松弛效应的主要组分。
9.4 抗惊厥作用 BDZ位点配体的抗惊厥作用只是部分地由含α1受体介导,除含α1受体外,其它GABAA受体亚型也起介导作用,BDZ的抗惊厥效应体现在其拮抗戊四氮所诱发的紧张性抽搐。α1[H101R]小鼠表现出BDZ抗惊厥效应,是由其他非α1受体的GABAA受体亚型介导的,因为该效应能被BDZ拮抗剂氟马西尼所拮抗。研究表明抗惊厥效应是部分而非完全由α1受体介导的。
9.5 失忆作用 BDZ诱发的顺行性遗忘也是由含α1受体介导的,这是因为α1[H101R]突变小鼠中BDZ的记忆损伤效应显著降低。
10 作用于GABAA受体的药物
GABAA受体是临床上广泛使用的神经药物的作用靶点,如镇静剂安定类、抑制剂巴比妥类、痉挛剂印防己毒素、乙醇(目前研究已证实,在basolateral锯齿状扁桃体脑回路径中,乙醇通过增强GABAA受体介导的神经传导来抑制长效势差现象产生的诱导作用,乙醇的这一作用与它所诱导的与情绪相关的学习和记忆损伤密切相关)、麻醉剂神经固醇类和植物活性成分(黄酮类、生物碱类、呋喃香豆精类和二萜醌类)等。
10.1 GABA结合位点的兴奋剂 GABA识别位点的兴奋剂除GABA外,有蝇蕈醇及其衍生物二氢蝇蕈醇及硫代蝇蕈醇,蝇蕈醇是研究GABA识别位点药理作用最广泛使用的外源性兴奋剂,其抑制作用较GABA强10倍,但毒性较大。
10.2 GABA结合位点的拮抗剂 GABA识别点的拮抗剂 Bic是从植物兜状荷包牡丹中首次分离而得到的异喹啉类生物碱,具有拮抗GABA引起的抑制效应,是GABAA受体的竞争性拮抗剂。此外还有合成的拮抗剂毗拉西平,取代蝇蕈醇与受体结合的能力较Bic强10倍,但特异性较差,在高质量分数时也可取代氟硝西泮与BDZ识别位点结合。
10.3 BDZ识别位点的兴奋剂、反相兴奋剂和拮抗剂 临床常用的地西泮类(BDZ类)药物包括地西泮、硝西泮、氯硝西泮、氟硝西泮、氯氮及三唑仑等均可与BDZ识别位点高亲和力稳定结合,都是BDZ识别位点的兴奋剂。BDZ 类通过促进GABA与GABAA受体的结合易化GABA功能,起到加强GABA的抑制效应,从而发挥其镇静、催眠、抗焦虑、中枢性肌肉松弛和抗惊厥作用。有人称BDZ类药物是GABA受体的正性变构调节物,不仅具有抗焦虑镇静剂和肌肉松弛剂的作用,同时具抑郁等副作用。由于BDZ类药物与BDZ受体结合没有选择性,因此在发挥抗焦虑和镇静催眠作用的同时,具有肌肉松弛、记忆力减退、耐受和成瘾性等副作用,使其应用受到很大限制。三唑哒嗪也是BDZ识别位点的兴奋剂,其增强GABA结合亲和力不如BDZ类药物强,但副作用相对小。在寻找作为GABAA受体正性变构调节物而起作用的、与BDZ识别位点结合的内源性配体时,在人尿中提取出了能与BDZ识别位点呈高亲和力结合,但作用效果与BDZ类相反,即拮抗GABA的抑制作用,具有致焦虑、致惊厥作用,故称之为反相兴奋剂。氟马西尼是BDZ识别位点的高效拮抗剂,能阻滞它们的正性、负性变构调节作用及其行为反应,但它本身没有作用。
10.4 作用于Cl-通道的药物 GABAA受体的另一组成部分是Cl-通道。Cl-通道也是药物作用的一个位点,有的药物不影响GABA与BDZ和受体识别位点结合的亲和性,而是直接影响Cl-通道,产生效应。如抑制剂巴比妥类通过延长每个通道启开的时间,除本身可产生超极化作用外,还可增强并延长GABA的超极化作用。而印防己毒素则关闭Cl-通道而阻滞GABA受体的作用。Bic和印防己毒素都能阻滞GABA的抗惊厥作用,但作用机理却完全不同。另外巴比妥能加强[3H]-GABA对大脑膜的结合,此作用可被印防己毒素抑制,但不被BDZ识别位点拮抗剂氟马西尼抑制。表明巴比妥类和印防己毒素有着相同或相近作用位点。神经活性甾体治疗一系列临床紊乱症状,如焦虑、癫痫和经前期综合征等,通过作用于Cl-通道而产生疗效。这类甾体按其功能可分为麻醉性甾体和兴奋性甾体,前者能导致催眠、镇痛、麻醉和抗惊厥等作用,后者能引发惊厥。兴奋性类固醇能通过减少通道启开频率来拮抗GABAA受体介导的Cl-电流,这种特异结合能被巴比妥抑制[17]。
10.5 巴比妥类(BB) 巴比妥类药物被广泛用作抗抽搐药、镇静催眠药和麻醉药,通常情况下,其对GABAA受体有以下3种不同的作用机理被人们接受:①加强GABA引发的电流反应;②直接激活GABAA受体通道;③在高质量分数情况下阻滞GABAA受体Cl-通道[18]。
10.6 其它作用于 GABAA受体的化合物 主要的还有麻醉剂、乙醇、不饱和的游离脂肪酸以及多价离子等。为了寻找高效低毒的新型神经活性药物,近年来应用GABAA/BDZ受体结合分析方法,从传统和民间药用植物中筛选活性部位,并进一步分离有效成分,取得了可喜进展,如:黄酮类、生物碱类等[2]。
11 展 望
随着分子生物学研究的深入及生物技术研究手段的发展,GABAA受体的结构及功能将逐渐被解释清楚,在此基础上,科学家将更深入了解GABAA受体各亚基的具体功能,加深对大脑功能包括癫痫在内的神经或精神疾病的病因学的理解,以期设计出新型有效的治疗策略,并将开发更具有针对性的神经调控药物。
参考文献:
[1]Krasowski MD, Kohchine VV, Rick CE, et al. Propofol and other intravenous anesthetics have sites of action on the gamma-aminobutyricacid type A receptor distinct from th at for isoflurane[J]. Mol Phammcol, 1998, 53(3): 530-538.
[2]王秀坤,Nielsen M.GABA_A受体药理学研究进展[J].国外医学:药学分册,2001,28(1):29-34.
[3]Thompson SA, Wafford K. Mechanism of action of general anaesthetics new information from molecu 2 lar pharmacology[J]. Curr Opin Pharmacol, 2001, 1(1): 78-83.
[4]Crestani F, Martin JR, Mohler H, et al. Mechanism of the hypnotic zolpidem in vivo[J]. Br J Pharmacol, 2000, 131(7): 1251-1254.
[5]Bormann J. The"ABC"of GABA receptors[J]. Trends Pharmacol Sci, 2000, 21(1): 16-19.
[6]Mehta AK, Ticku MK. An update on GABAA receptors[J]. Brain Res Rev, 1999, 29(2-3): 196-217.
[7]张瑞华,李丽琴,王惠芳.GABAA受体药理学研究进展[J].中国药理通讯,2003,20(3):18-19.
[8]Liu M, Glowa JR. Regulation of benzodiazepine receptor binding and GABA(A)subunit mRNA expression by punishment and acute alprazolam administration[J]. Brain Res, 2000, 887: 23-33.
[9]Rudolph U, Crestani F, Möhler HG. (A)receptor subtypes:dissecting their pharmacological functions[J]. Trends Pharmacol Sci, 2001, 22(4): 188-194.
[10]Corringer PJ, Le NN, Changeux JP. Nicotinic receptors at the amino acid level[J]. Annu Rev Pharmacol Toxicol, 2000, 40(1): 431-458.
[11]Zhang DX, Pan ZH, Marc A, et al. Structure and function of GABAC receptors:a com 2 parison of native versus recombinant receptors[J]. Trends Pharmacol Sci, 2001, 22(3): 121-132.
[12]SubramaniamJR, Corsi C, Vicini S, et al. Ribozyme-mediated reduction of the GABAA receptor α1 subunit[J]. Molecular Brain Research, 2001, 92(1-2): 149-156.
[13]小川纪雄原.脑受体[M].北京:北京医科大学、中国协和医科大学联合出版社,1997:322-324.
[14]Wingrove PB, Thompson SA, Wafford KA, et al. Key amino acids in the subunit of the aminobutyric acidA receptor that determine ligand binding and modulation at the benzodiazepine site[J]. Mol Pharmacol, 1997, 52(5): 874-881.
[15]郭文,王丽.氯巴占耐受听源性惊厥大鼠脑GABAA受体亚单位表达的变化[J].中国药理学通报,2002,18(2):183-186.
[16]Rudolph U, Crestani F, Bebe D, et al. Benzodiazepine actions mediated by specific gamma aminobutyric acid A receptor subtypes[J]. Nature, 1999, 401: 796-800.
[17]柳海珍,朱剑琴.神经活性甾体与GABA_A受体[J].生命科学,1997,9(4):162-165, 157.
[18]Krampfl K, Wolfes H, Dengler R, et al. Kinetic analysis of the agonistic and blocking properties of pentobarbital on recombinant rat alpha(1)beta(2)gamma(2S)GABA(A)receptor channels[J]. European J Pharmacology, 2002, 435(1): 1-8.
猜你喜欢
世界最新医学信息文摘(2020年68期)2020-12-25
延安大学学报(医学科学版)(2019年1期)2019-03-29
中成药(2018年9期)2018-10-09
中成药(2018年8期)2018-08-29
中西医结合心脑血管病杂志(2016年20期)2016-03-01
中国继续医学教育(2015年6期)2016-01-07
中国病理生理杂志(2015年8期)2015-12-21
中国当代医药(2015年30期)2015-03-01
中国民族民间医药·下半月(2014年4期)2014-09-26
浙江中西医结合杂志(2014年12期)2014-01-23
