
无溶剂条件下新型N-羟乙基-9-取代十氢吖啶-1,8-二酮衍生物的合成
2014-08-05 04:42:39常州大学精细化工重点实验室江苏常州213164
合成化学 2014年1期
(常州大学 精细化工重点实验室,江苏常州 213164)
(常州大学 精细化工重点实验室,江苏常州 213164)
通过芳香醛、达米酮和乙醇胺的“一锅法”无溶剂反应将羟乙基引入吖啶二酮的氮原子上,合成了一系列新型的吖啶二酮衍生物,收率29%~83%,其结构经1H NMR,13C NMR,IR和MS表征。并对其反应机理进行推测。
芳醛;达米酮;乙醇胺;吖啶二酮;“一锅法”;无溶剂合成
1,4-二氢吡啶(DHP)类化合物可用于治疗心率不调[1],是一类重要的钙离子通道阻滞剂[2-4]。在DHP上引入不同的取代基或杂原子可制得不同生物活性物质。已经证实在DHP环上稍作修饰便会带来不同的药理作用[5]。多氢吖啶衍生物含有1,4-DHPs母核,已经发现其具有抗菌、抗疟疾及抗癌等作用,多种药物是以多氢吖啶为母体合成的[6-9]。9-芳基-十氢吖啶-1,8-二酮具有1,4-二氢吡啶结构单元,可用作ATP钾离子通道调节剂[10-11]。另外,吖啶二酮可作为一种激光染料[12],其具有双重发色结构,在基态和激发态时均既可作为电子接受体也可作为电子给予体[13];在光聚合反应中作为光敏剂和光引发剂也具有潜在的应用[14],而且还应用于在染料光化学[15]和光物理[16-17]等领域。
随着绿色化学的发展,十氢吖啶-1,8-二酮化合物[18-19]的合成可利用水相、微波辐射及无溶剂合成等方法。本课题组[20]曾用无溶剂及水相方法合成了十氢吖啶-1,8-二酮类化合物。屠树江等[21]报道了在微波条件下醛肟和双分子达米酮或1,3-环己二酮反应合成了一系列新型的N-羟基-9-芳基十氢吖啶-1,8-二酮衍生物。然而,人们对于N上含有末端羟基取代的9-芳基-十氢吖啶-1,8-二酮报道甚少[21]。我们试图找到一种合成氮原子上含有末端羟基取代的9-芳基-十氢吖啶-1,8-二酮类化合物的方法,以期获得新的活性物质。

Scheme 1
本文通过达米酮(1)、芳香醛(2a~2o)和乙醇胺的“一锅法”无溶剂反应将羟乙基引入吖啶二酮的氮原子上,合成了一系列新型的吖啶二酮衍生物(3a~3o,Scheme 1),其结构经1H NMR,13C NMR,IR和MS表征。并对其反应机理进行推测。
1 实验部分
1.1 仪器与试剂
XT4-100X型显微熔点仪(温度未校正);AVANCE 500 MHz型核磁共振仪(DMSO-d6为溶剂,TMS为内标);Nicolet PROTÉGÉ 460型红外光谱仪(KBr压片)。
所用试剂均为分析纯。
1.2 3a~3o合成(以3a为例)
在试管中加入苯甲醛(2a)140 mg(1 mmol),1 280 mg(2 mmol)和乙醇胺67 mg(1.1 mmol)。将试管置于经预热的油浴(160℃),搅拌(反应物立刻熔化为黄色液体)反应(溶液颜色加深呈深褐色,并且变粘稠)50 min(TLC监测)。加入DMF 2 mL~3 mL,加热使其溶解;搅拌下倒入冰水中(大量固体析出)。抽滤,滤饼干燥得淡黄色固体3a 290.3 mg,产率71%。用乙醇重结晶可提高其纯度。
用类似方法合成淡黄色固体3b~3o(3n和 3o:加入冰水后,先用CH2Cl2萃取,再用乙酸乙酯/石油醚重结晶)。
2 结果与讨论
2.1 合成
在3a的合成中,对反应温度进行考察。2a 1 mmol,其余反应条件同1.2,考察温度(100℃,120℃,140℃和160℃)对反应的影响。结果表明,于160℃反应60 min后,TLC监测结果显示原料2a和1已经完全消失,生成了一个荧光很强的单一产物。而在100℃,120℃和140℃反应,效果较差,从TLC显示该反应生成了比较多的副产物,其中主要副产物为2'-(芳基亚甲基)-3-羟基-2-环己烯-1-酮,并且反应时间延长,不能生成单一产物3a。由此推断该反应在高温(160℃)条件下较有利。
在无溶剂反应过程中,反应混合物首先转变为均一的液相,随后很快反应转化为固体产物。Scott等[22-23]举例说明了无溶剂反应高效的一个主要原因是形成了一个较低共熔点,可以在很高的浓度下进行反应。在本研究中,所有原料的熔点都低于160℃,因此形成的均一液相可能是简单的熔化亦或产生了一个共熔点,致使所有原料达到较高浓度,从而使得反应比在有机溶剂中速度更快、产率更高。
鉴于上述实验结果,对底物进行扩展,尝试了其它醛类化合物(2b~2f)与1及乙醇胺在无溶剂条件下于160℃反应,均获得了N-羟乙基-9-芳基十氢吖啶-1,8-二酮衍生物(3b~3f),实验结果见表1。由表1可见,大部分底物反应产率均比较理想(>70%);苯环上取代基的电子效应对该反应无影响;以N,N-二甲基苯甲醛为底物时,产率较低(29%),因为其在高温条件下容易被氧化,副反应较多;以叔丁基醛(2n)或乙基醛(2o)为底物时,产率只有31%和36%,副产物较多。
2.2 表征
3a~3o的实验结果,IR和MS数据见表1,NMR数据见表2。
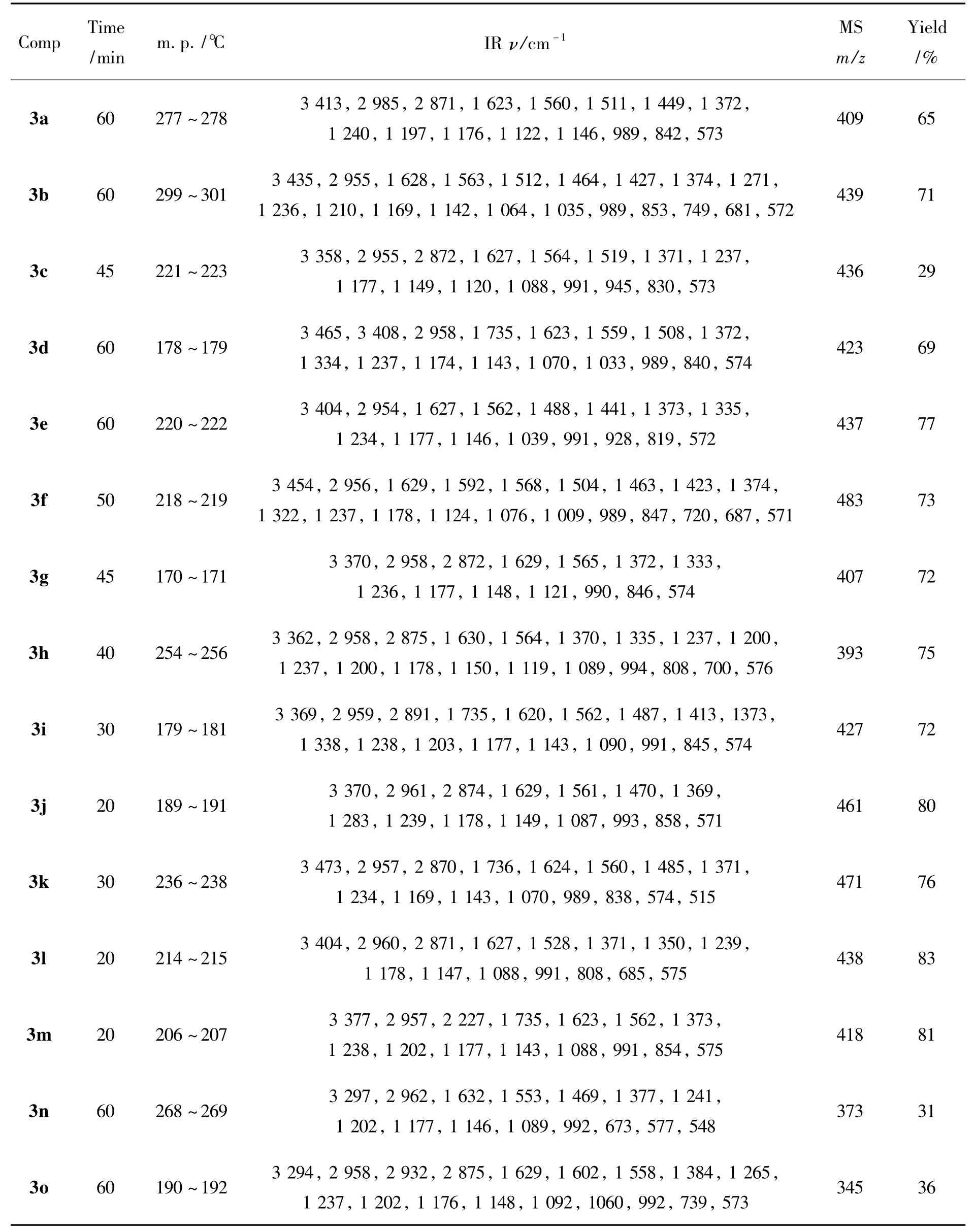
表1 3a~3o的实验数据,IR和MS数据Table 1 Experimental results,IR and MS data of 3a~3o

表2 3a~3o的NMR数据Table 2 NMR data of 3a~3o

续表2
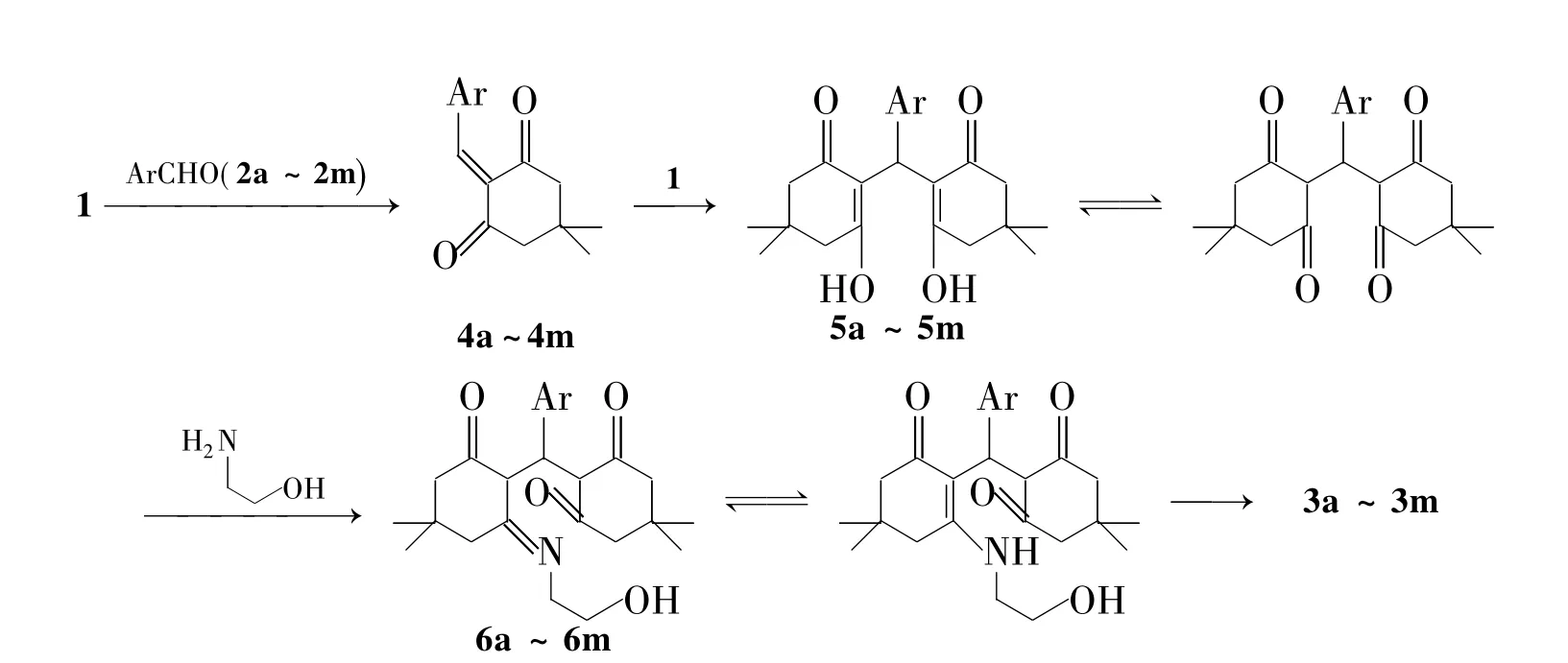
Scheme 2
1H NMR分析结果表明,在3h中,1.01和1.08处出现达米酮骨架上两个不等性甲基的六个氢信号;在2.26~2.65共出现了8个氢信号,归属4个CH2氢质子;在3.71和3.87处出现了两个三重峰,有四个氢,归属羟乙基上的两个亚甲基质子;在5.34处出现的单峰为9-位与芳基相连的CH信号;低场区域7.25处的双峰,7.17处的三重峰和7.07处的三重峰为苯环上的氢质子;在1.76处出现的宽峰为羟基信号。
在3a的IR谱中,3 361 cm-1处出现了明显的OH特征伸缩振动吸收峰。
2.3 反应机理
我们提出了3a~3m的形成机理(Scheme 2)。首先2a~2m和1缩合原位生成4a~4m;随后和1发生迈克尔加成反应得中间体5a~5m,接着和乙醇胺发生缩合,再异构化为6a~6m,然后通过分子内加成、关环脱水得最终吖啶二酮3a~3m。
3 结论
以芳香醛、达米酮和乙醇胺为原料,通过尝试在无溶剂、水相、有机溶剂相的无溶剂“一锅法”反应,创建了一种合成N-(2-羟基)乙基-9-芳基-十氢吖啶-1,8-二酮类化合物的良好方法。该方法直接采用高温(160℃)下无溶剂反应,所得固体用乙醇加热溶解,趁热过滤,通过冷却和溶剂挥发结晶,获得纯度较高的产品。该反应过程和处理方法比较环保,产率较高。
该类氮原子上含有末端羟乙基取代的9-芳基-十氢吖啶-1,8-二酮化合物可以通过醚化、酯化等多种方式对羟基加以修饰,预期可获得多样修饰的十氢吖啶-1,8-二酮衍生物。
[1]Bossert F,Meyer H,Wehinger E.4-Aryldihydropyridines,a new class of highly-active calcium-antagonists[J].Angew Chem Int Ed Eng,1981,20:762-769.
[2]Stout D M,Meyers A I.Recent advances in the chemistry of dihydropyridines[J].Chem Rev,1982,82: 223-243.
[3]Janis R A,Triggle D J.New developments in ca-2+ channel antagonists[J].J Med Chem,1983,26: 775-785.
[4]Kappe C O.100 Years of the biginelli dihydropyrimidine synthesis[J].Tetrahedron,1993,49:6937-6963.
[5]Schramm M,Thomas G,Tower R,et al.Novel dihydropyridines with positive inotropic action through activation of ca-2+ channels[J].Nature,1983,303: 535-537.
[6]Wysockaskrzela B,Ledochowski A.Research on tumor inhibiting compounds.55.Syntheses of n-substituted 4-nitroacridone,and 1-nitro-4-methylacridone derivatives[J].Roccz Chem,1976,50:127-131.
[7]Nasim A,Brychcy T.Genetic-effects of acridine compounds[J].Muta Res,1979,65,261-288.
[8]Thull U,Testa B.Screening of unsubstituted cycliccompounds as inhibitors of monoamine oxidases[J]. Biochem Pharmacol,1994,47:2307-2310.
[9]Reil E,Soll M,Masson K,et al.Synthesis of quinolones and acridones and their inhibitory activity in nadhdehydrogenases and cytochrome bc(1)-complexes[J]. Biochem Soc Trans,1994,22:S62-S62.
[10]Simsek R,Ozkan M,Kismetli E,et al.Some ary-lacridine derivatives possessing potassium channel opening activity[J].Farmaco,2004,59:939-943.
[11]Gopalakrishnan M,Miller T R,Buckner S A,et al.Pharmacological characterization of a 1,4-dihydropyridine analogue,9-(3,4-dichlorophenyl)-3,3,6,6-tetramethyl-3,4,6,7,9,10-hexahydro-1,8(2H,5H)-acridinedione(A-184209)as a novel K-ATP channel inhibitor[J].Br J Pharmacol,2003,138:393-399.
[12]Shanmugasundaram P,Murugan P,Ramakrishnan V T,et al.Synthesis of acridinedione derivatives as laser dyes[J].Heteroatom Chem,1996,7:17-22.
[13]Mohan H,Srividya N,Ramamurthy P,et al.One-electron reduction of acridine-1,8-dione in aqueous solution:A pulse radiolysis study[J].J Phys Chem,1997,101:2931-2935.
[14]Timpe H J,Ulrich S,Decker C,et al.Photoinitiated polymerization of acrylates and methacrylates with decahydroacridine-1,8-dione/onium salt initiator systems[J].Macromolecules,1993,26:4560-4566.
[15]Islam A,Murugan P,Hwang K C,et al.Blue light-emitting devices based on 1,8-acridinedione derivatives[J].Synthetic Metals,2003,139:347-353.
[16]Srividya N,Ramamurthy P,Ramakrishnan V T.Photophysical studies of acridine(1,8)dione dyes:A new class of laser dyes[J].Spectrochim Acta,1998,54 (A):245-253.
[17]Srividya N,Ramamurthy P,Ramakrishnan V T.Solvent effects on the absorption and fluorescence spectra of some acridinedione dyes:Determination of ground and excited state dipole moments[J].Spectrochim Acta,1997,53(A):1743-1753.
[18]Jin T S,Zhang J S,Guo T T,et al.One-pot clean synthesis of 1,8-dioxo-decahydroacridines catalyzed by p-dodecylbenezenesulfonic acid in aqueous media[J].Synlett,2004,2001-2005.
[19]Tu S-J,Lu Z,Shi D,et al.A convenient synthesis of 9-aryl-3,3,6,6-tetramethyl-1,2,3,4,5,6,7,8,9,10-decahydroacridine-1,8-diones under microwave irradiation without solvent[J].Synth Commun,2002,32: 2181-2185.
[20]Wang G,Miao C.Environmentally benign one-pot multicomponent approaches to the synthesis of novel unsymmetrical 4-arylacridinediones[J].Green Chem,2006,8:1080-1085.
[21]Tu S,Miao C,Gao Y,et al.A Novel cascade reaction of aryl aldoxime with dimedone under microwave irradiation:The synthesis of N-hydroxylacridine[J]. Synlett,2004,255-258.
[22]Rothenberg G,Downie A P,Raston C L,et al.Understanding solid/solid organic reactions[J].J Am Chem Soc,2001,123:8701-8708.
[23]Cave G W V,Raston C L,Scott J L.Recent advances in solventless organic reactions:Towards benign synthesis with remarkable versatility[J].Chem Commun,2001,2159-2169.
无溶剂条件下新型N-羟乙基-9-取代十氢吖啶-1,8-二酮衍生物的合成*
董春萍,缪春宝,杨海涛,孙小强
Solvent-free Synthesis of Novel N-(2-hydroxyl)ethyl-9-subsituted-decahydroacridinedione Derivatives
DONG Chun-Ping, MIAO Chun-bao, YANG Hai-tao, SUN Xiao-qiang
(Key Laboratory of Fine Petrochemical Technology,Changzhou University,Changzhou 213164,China)
A series of novel 9-subsituted-decahydroacridine-1,8-dione derivatives in yield of 29% ~83%were synthesized by“one-pot”method of aromatic aldehydes,dimedione and ethanol amine under solvent-free conditions.The structures were characterized by1H NMR,13C NMR,IR and MS.The reaction mechanism was proposed.
aromatic aldehyde;decahydroacridine;ethanol amine;acridinedione;“one-pot”;solventfree synthesis
O626.32
A
1005-1511(2014)01-0024-06
2012-11-05;
2013-12-16
国家自然科学基金资助项目(20902039,21202011)
董春萍(1989-),女,汉族,江苏盐城人,主要从事多肽合成的研究。
缪春宝,副教授,E-mail:chunbao@cczu.edu.cn
猜你喜欢
农产品加工(2021年8期)2021-05-20 01:38:28
杭州师范大学学报(自然科学版)(2016年2期)2016-05-05 03:22:14
绥化学院学报(2016年3期)2016-03-23 09:00:14
当代化工研究(2016年7期)2016-03-20 16:21:54
西北师范大学学报(自然科学版)(2015年6期)2016-01-19 01:55:14
船舶标准化工程师(2015年5期)2015-12-03 11:00:24
中国高新技术企业(2015年13期)2015-04-30 21:07:38
西部皮革(2015年15期)2015-02-28 18:14:36
科技致富向导(2013年11期)2013-07-05 05:27:52
石油化工(2013年3期)2013-05-03 01:54:14
