
2,2-二丙基-β-丙氨酸的合成
2014-08-05 04:42:57四川大学生物治疗国家重点实验室四川成都610041中国科学院成都有机化学研究所四川成都610041
合成化学 2014年1期
(1.四川大学生物治疗国家重点实验室,四川成都 610041;2.中国科学院成都有机化学研究所,四川成都 610041)
(1.四川大学生物治疗国家重点实验室,四川成都 610041;2.中国科学院成都有机化学研究所,四川成都 610041)
以氰基乙酸乙酯为起始原料,在强碱作用下与溴丙烷经两次烷基化反应制得2-氰基-2-丙基-戊酸乙酯(3);3中氰基经镍催化还原为氨基得2-氨甲基-2-丙基-戊酸乙酯(4);4经水解反应合成了2,2-二丙基-β-丙氨酸,总收率37.4%,其结构经1H NMR确证。
氰基乙酸乙酯;β-氨基酸;烷基化;合成
氨基酸及其衍生物具有与生物体内活性物质相同或相似的基本组成,易被生物体吸收利用,已被越来越多地应用于化妆品、农药、医药、保健品等领域。与α-氨基酸相比,β-氨基酸分子中氨基和羧基之间多了一个碳原子,并增加了一个侧链,具有更多的结构多样性。β-氨基酸是β-内酯和内酰胺[1]的前体、天然产物及合成改性肽链的重要结构单元[2],如Merck公司研制已上市的抗高血糖药物Januvia[3]和Sanafi aventis公司研制的抗肿瘤药物Taxotere[4]中均含有β-氨基酸片段。目前,随着组合化学在氨基酸类新药、饲料及化妆品类添加剂合成方面的应用,需要大量新型β-氨基酸作为构建肽化合物库的基本单元,因此β-氨基酸的合成研究有广泛的实际应用价值。
目前β-氨基酸的合成方法主要有 Rodionov法[5],Arndt-Eistert法[6]和 Kibayashi法[7]等。本文以氰基乙酸乙酯(1)为起始原料,在强碱作用下与溴丙烷经两次烷基化反应制得2-氰基-2-丙基-戊酸乙酯(3);3中氰基经Raney Ni催化还原为氨基得2-氨甲基-2-丙基-戊酸乙酯(4);4经水解合成了2,2-二丙基-β-丙氨酸(5,Scheme 1),总收率37.4%,其结构经1H NMR确证。
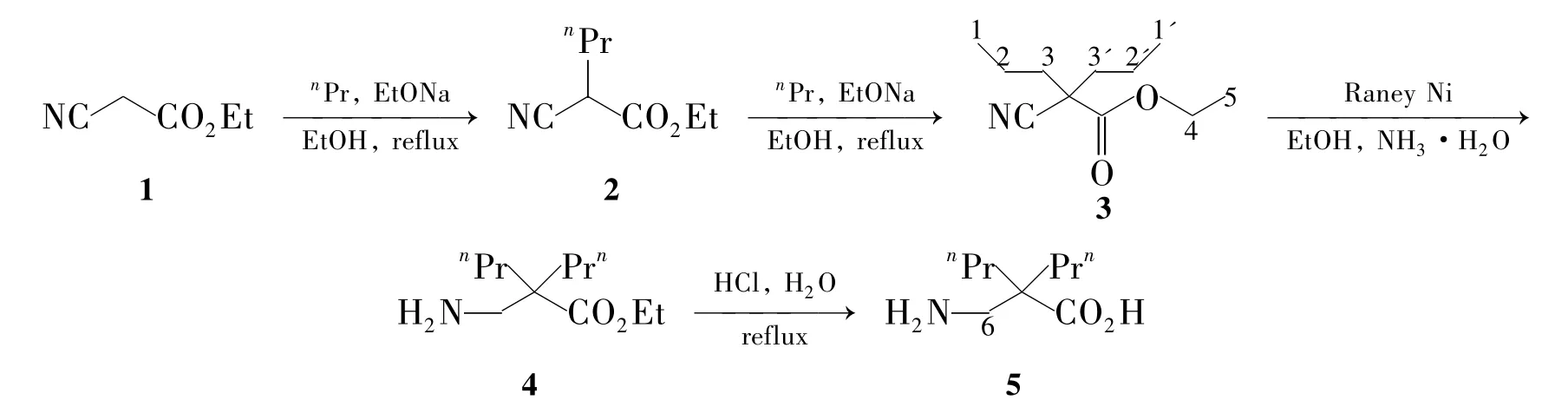
Scheme 1
1 实验部分
1.1 仪器与试剂
Bruker DRX-300 MHz型核磁共振仪(CDCl3为溶剂,TMS为内标)。
所用试剂均为化学纯,其中乙醇经镁条回流后蒸馏除水。
1.2 合成
(1)2-氰基-戊酸乙酯(2)的合成[8]
冰浴冷却下在反应瓶中依次加入绝对乙醇33 mL和金属钠1.3 g(56.5 mmol),搅拌下反应至钠块反应完毕;缓慢滴加1 2 mL(18.8 mmol),滴毕,回流反应20 min;冷却至室温,缓慢滴加溴丙烷3.4 mL(37.6 mmol),滴毕,回流反应8 h(体系由澄清变为红棕色悬浊液)。冷却至室温,旋蒸脱溶,加最少量水溶解,用DCM萃取,合并萃取液,用无水硫酸钠干燥,减压浓缩得红棕色液体2 2.39 g(直接进行下步反应);1H NMR δ:0.96 (t,J=7.32 Hz,3H,1-H),1.32(t,J=7.14 Hz,3H,5-H),1.52~1.58(m,2H,2-H),1.89~1.97(m,2H,3-H),3.47~3.51(m,1H,CH),4.27(q,2H,J=7.14 Hz,4-H)。
(2)3的合成
冰浴冷却下在反应瓶中依次加入绝对乙醇33 mL和金属钠1.3 g(56.5 mmol),搅拌下反应至钠块反应完毕;缓慢滴加2 2.39 g,滴毕,回流反应20 min;冷却至室温,缓慢滴加溴丙烷3.4 mL(37.6 mmol),滴毕,回流反应8 h(体系由澄清变为红棕色悬浊液)。冷却至室温,减压浓缩后用最少量水溶解,用DCM萃取,合并萃取液,用无水硫酸钠干燥,旋蒸脱溶后经硅胶柱层析[洗脱剂:V(EA)∶V(PE)=20∶1]纯化得淡黄色液体3 1.93 g,两步总收率51.2%;1H NMR δ: 0.92(t,J=7.32 Hz,6H,1,1'-H),1.27~1.33 (m,5H,2,5-H),1.70~1.72(m,2H,3-H),1.74~1.76(m,2H,3'-H),1.84~1.85(m,2H,3'-H),4.23(q,J=7.14 Hz,2H,4-H)。
(3)4的合成
在高压釜中依次加入3 0.5 g(2.53 mmol),乙醇10 mL,氨水5 mL及Raney Ni 0.5 g(湿重),用H2置换6次,通H2至4 MPa,于40℃反应48 h。冷却至室温,过滤,滤液浓缩后用DCM萃取,合并萃取液,用水洗涤,用无水硫酸钠干燥,浓缩得淡黄绿色黏稠液体4 0.4 g,产率78.6%;1H NMR δ:0.89(t,J=7.20 Hz,6H,1,1'-H),1.17~1.26(m,7H,2,2',5-H),1.48~1.54 (m,4H,3,3'-H),2.79(s,2H,6-H),4.12(q,J=7.14 Hz,2H,4-H)。
(4)5的合成
在反应瓶中依次加入4 1 g(4.97 mmol)和6 mol·L-1HCl溶液16.6 mL(99.4 mmol),回流反应4 h。冷却至室温,减压蒸干溶剂,残余物用少量乙酸乙酯洗涤得白色固体 5 0.8 g,产率92.9%;1H NMR δ:0.88(t,J=7.20 Hz,6H,1-H),1.15~1.54(m,4H,2,2'-H),1.34~1.63 (m,4H,3,3'-H),3.18(s,2H,6-H)。
2 结果与讨论
2.1 合成
(1)3合成
鉴于3的α-位是两个对称的烷基,我们曾试图通过采用强碱NaH拔除α-位的两个质子,同时加大卤代烷及碱用量,在1的α-位一次性实现双烷基化。但实验结果显示NaH为碱,无水THF为溶剂,室温及加热回流反应都很难进行完全,反应48 h后仍有大量1没有反应。即使当NaH及溴丙烷大大过量、多次添加,1仍有大量剩余,3的量很少,大部分为单取代产物2,使用相转移催化剂结果也不理想。改用醇钠为碱,在溴丙烷过量(4 eq~6 eq)条件下仍然生成2和3的混合物,2占主要成份,但反应进行较快,反应进行24 h后,反应基本完全。
由于一锅反应不能很好实现双烷基化,本文采用分步烷基化法。首先,在乙醇钠作用下拔除α-位质子,为防止酯交换引起产物复杂化,难以监测反应进程,采用乙醇为溶剂。无需加相转移催化剂,1与2 eq溴丙烷反应形成2与3的混合物,2无需分离纯化,反应结束后经简单处理后即可直接进行下步烷基化反应,顺利获得3,两步总收率51.2%。
(2)4合成
在4的合成中,氰基的还原反应使用Raney Ni为催化剂,催化剂用量(湿重)为底物质量一倍,同时采用混合溶剂[V(乙醇)∶V(氨水)= 2∶1]。加入氨水是为了防止生成还原产物的二聚体。为了避免3氨解,反应温度不宜过高,于40℃反应2 d反应进行完全。
(3)5合成
实验中首先尝试4在碱性条件下进行水解,即使加入甲醇或THF增溶,回流过夜也难以反应完全,且产物复杂,可能是分子内或分子间氨基与酯基发生反应所致。本文采用酸性条件下水解,质子酸与氨基形成盐保护了氨基,避免酰胺的形成导致产物复杂化。在酸性(6 mol·L-1HCl溶液)条件下回流反应4 h,反应体系为均相,水解顺利进行,蒸干溶剂即得5。
综上所述,该合成方法除3需柱层析纯化外,其余反应均无需柱层析或重结晶,简单处理后即得较高纯度产物。该方法可推广到单取代,对称双取代及非对称双取代β-氨基酸及其衍生物的合成研究之中。
[1]Alcaide B,Almendros P,Aragoncillo C.β-Lactams: Versatile building blocks for the stereoselective synthesis of non-β-lactam products[J].Chem Rev,2007,107 (11):4437-4492.
[2]Cheng R P,Gellman S H,DeGrado W F.β-Peptides: From structure to function[J].Chem Rev,2001,101 (10):3219-3232.
[3]Hansen K B,Hsiao Y,Xu F,et al.Highly efficient asymmetric synthesis of Sitagliptin[J].J.Am Chem Soc,2009,131(25):8798-8804.
[4]Guenard D,Guenritte-Voegelein F,Potier P.Taxol and taxotere:Discovery,chemistry,and structure-activity relationships[J].Acc Chem Res,1993,26:160-167.
[5]Rodionov W M.Synthesis of beta-aryl-amino-ethanealpha,alpha-dicarbonic acids the mechanism of Knovenagel's synthesis of cinnamic acids[J].J Am Chem Sci,1929,51(3):847-852.
[6]Plucinska K,Liberek B.Synthesis of diazoketones derived from α-amino acids;Problem of side reactions[J].Tetrahedron,1987,43(15):3509-3517.
[7]Kaseda T,Kikuchi T,Kibayashi C.Enanticselective total synthesis of(+)-(S)-dihydroperiphylline[J]. Tetrahedron Lett,1989,30(34):4539-4542.
2,2-二丙基-β-丙氨酸的合成*
许景刚1,李桃桃2,赵玉燕2,吴德志2,张 振2,支永刚2,余洛汀1
Synthesis of 2,2-Dipropyl-β-alanine
XU Jing-gang1, LI Tao-tao2, ZHAO Yu-yan2,WU De-zhi2, ZHANG Zhen2, ZHI Yong-gang2, YU Luo-ting1
(1.State key laboratory of biotherapy,Sichuan University,Chengdu 610041,China;2.Chengdu Institute of Organic Chemistry,Chinese Academy of Sciences,Chengdu 610041,China)
Ethyl 2-cyano-2-propylvalerate(3)was obtained from ethyl cyanoacetate by two-step alkylation.Ethyl 2-amino-2-propylvalerate(4)was prepared by catalytic reduction of 3 with Raney Ni. 2,2-Dipropyl-β-alanine was synthesized by hydrolysis of 4.The total yield was 37.4%.The structures were confirmed by1H NMR.
ethyl cyanoacetate;β-amino acid;alkylation;synthesis
O623.736
A
1005-1511(2014)01-0108-03
2013-10-30;
2013-12-16
许景刚(1977-),男,汉族,重庆人,硕士研究生,主要从事药物合成及生物催化的研究。E-mail:xujinggang@126.com
支永刚,研究员,博士导师,Tel.028-85229766,E-mail:zhiyonggang@hotmail.com
猜你喜欢
昆明医科大学学报(2021年2期)2021-03-29 07:42:14
四川警察学院学报(2019年6期)2019-12-28 07:20:06
中成药(2018年6期)2018-07-11 03:01:28
中国有色金属学报(2018年2期)2018-03-26 07:58:48
石油石化绿色低碳(2018年5期)2018-03-20 04:41:21
中成药(2017年5期)2017-06-13 13:01:12
文化产业(2016年6期)2016-10-19 19:13:47
石油炼制与化工(2016年6期)2016-04-06 22:54:21
合成化学(2015年2期)2016-01-17 09:04:08
合成化学(2015年9期)2016-01-17 08:57:14
