
人端粒酶逆转录酶基因小片段干扰RNA慢病毒表达载体的构建及其在子宫颈癌caski细胞的表达
2014-07-01 22:15赵红珂莫凌昭
中国癌症防治杂志 2014年4期
赵红珂莫凌昭
作者单位:530021 南宁 广西医科大学附属肿瘤医院妇瘤科;△广西医科大学研究生学院
基础研究
人端粒酶逆转录酶基因小片段干扰RNA慢病毒表达载体的构建及其在子宫颈癌caski细胞的表达
赵红珂△莫凌昭
作者单位:530021 南宁 广西医科大学附属肿瘤医院妇瘤科;△广西医科大学研究生学院
目的 构建及鉴定携带沉默人端粒酶逆转录酶基因(human telomerase reverse transcriptase,hTERT)慢病毒(lentivirus,LV)表达载体,并观察其在子宫颈癌caski细胞的表达情况。方法 以Designer3.0(Genepharma)软件设计靶向hTERT基因特异性的小片段RNA(shRNA)干扰序列,将hTERT-shRNA基因片段插入重组慢病毒pGLV3/H1/GFP+Puro,构建慢病毒表达质粒LV3-shRNA-hTERT,酶切、DNA测序验证hTERT片段准确性。LV3-shRNA-hTERT与包装质粒共转染293T细胞,浓缩上清液并测定病毒滴度获得重组慢病毒后感染caski细胞。将细胞分为空白对照组(未感染病毒的caski细胞)、阴性对照组(感染空载病毒LV3-shNC的caski细胞)和hTERT干扰组(感染携带LV3-shhTERT慢病毒的caski细胞)。绿色荧光蛋白(GFP)的表达判断转染结果并估计转染效率,流式细胞术仪检测病毒感染率,实时荧光定量PCR法(qRT-PCR)检测转染后基因hTERT mRNA的表达情况,CCK-8法检测caski细胞增殖情况。结果 将目的序列成功连接到载体上,并经测序分析证实载体构建成功;成功包装成高滴度的慢病毒,且能有效感染子宫颈癌caski细胞。荧光定量PCR检测结果证实构建的hTERT-小片段干扰RNA慢病毒表达载体可显著抑制hTERT基因的表达。hTERT干扰组细胞增殖受抑制,生长速度较阴性对照组生长缓慢。结论 成功构建了携带hTERT-shRNA慢病毒表达载体LV3-shRNA-hTERT,并稳定转染子宫颈癌caski细胞。该载体能够有效抑制hTERT的表达,使子宫颈癌caski细胞增殖缓慢,为进一步探讨hTERT基因在子宫颈癌的发病机制和体外基因干预治疗奠定基础。
子宫颈肿瘤;人端粒酶逆转录酶基因;慢病毒载体;小片段RNA;caski细胞
子宫颈癌是常见的女性恶性肿瘤之一,其发病率居于我国女性生殖系统恶性肿瘤的首位,发展中国的家的发病率高于发达国家[1]。调查显示,子宫颈癌的发生和发展过程中高危型人乳头状瘤病毒,如HPV16及HPV18等病毒的持续感染为重要的关键致病因素[2]。高危型HPV E6蛋白诱导并维持子宫颈癌细胞的端粒酶表达[3],可见端粒酶为一种重要的靶蛋白。端粒酶是一种细胞RNA依赖的DNA聚合酶,能够维持细胞染色体末端端粒长度。调节端粒酶活性的限速因素是端粒酶的催化亚单位—人体端粒酶逆转录酶(human telomerase reverse transcriptase,hTERT)[4]。端粒酶作为恶性肿瘤标志物已被认可,但端粒酶抑制剂可否作为抗肿瘤药物仍未知,以端粒酶为靶点筛选新的抗肿瘤药物成为目前抗肿瘤研究的热点之一[5]。RNA干扰(RNA interference,RNAi)是小片段干扰 RNA(small interference RNA,siRNA)在转录后mRNA水平关闭靶基因表达的现象[6]。干扰hTERT基因在子宫颈癌caski细胞的表达能否作为子宫颈癌基因治疗的一个新靶点有待研究。本研究利用RNAi技术,构建干扰hTERT基因重组慢病毒表达质粒,并将其转入该基因高表达的子宫颈癌caski细胞中,进一步观察hTERT对肿瘤细胞增殖的影响,以期为研究hTERT与子宫颈癌的发病机制提供理论基础。
1 材料与方法
1.1 主要试剂与材料
子宫颈癌caski细胞株和人胚胎肾上皮293T细胞购自中国科学院细胞库,感受态大肠杆菌DH-5α、质粒 pGLV3/H1/GFP+Puro、Polybrene和 RNAi-Mate转染试剂购自上海吉玛基因有限公司。限制性内切酶和T4DNA连接酶购自日本TaKaRa公司,Trizol购自美国Sigma公司,逆转录试剂盒购自美国Invitrogen公司,RPMI1640培养基、DMEM培养基和胎牛血清购自美国Hyclone公司,PCR引物合成、测序、慢病毒重组穿梭质粒和包装质粒pGag/Pol、pRev、pVSV-G的制备均为上海吉玛基因有限公司。
1.2 表达hTERT-shRNA质粒载体的构建
1.2.1 hTERT-shRNA的设计 根据 GeneBank中hTERT的mRNA序列(NM198253),参照siRNA靶点设计原则[7],应用siRNA靶点设计软件Oligo Designer3.0(Genepharma),设计并筛选出一个有效的靶位点:GCTTCCTCAGGAACACCAAGA,另行设计一条无效序列:TTCTCCGAACGTGTCACGT,对所选序列进行生物信息学分析(Blast分析、SNP分析)。
1.2.2 慢病毒载体的构建及包装 ⑴LV3-hTERT-shDNA模板的退火。将DNA oligo分别用TE(pH8.0)溶解,浓度为100 μmol/L。取相应的正义链和反义链
oligo溶液,按照配比配置退火反应体系。⑵LV3载体的线性化。取10 μg LV3载体,37℃酶切1 h,电泳后DNA凝胶回收试剂盒回收,使用紫外分光光度计测定浓度。⑶LV3-hTERT-shDNA载体的构建。反应体系10×T4连接缓冲液2 μl,载体LV3(BamHI+EcoRI)1 μl,shDNA模板(100 μmol/L)1 μl,T4 DNA连接酶(5 weissU/μl)1 μl,水15 μl,Total 20,22℃1 h,进行载体连接。⑷感受态细胞的制备,用氯化钙法。⑸连接产物的转化。解冻后的感受态细胞加10 μl连接产物,冰敷30 min。42℃水浴90 s,冷却3 min。加入800 μl LB培养基,37℃摇床,250 r/min离心45 min后细菌复苏。取200 μl培育后的细胞均匀涂布于含50 μg/ml Ampi-cillin LB平板上。倒置平板于37℃培养16 h。⑹阳性克隆的鉴定与测序。使用碱裂解法抽提质粒,所得质粒用EcoRI进行单酶切鉴定,将阳性克隆进行测序鉴定。
1.2.3 慢病毒浓缩和收集 对293T细胞进行常规培养,细胞密度达85%~95%时进行细胞传代,接种于3个15 cm培养皿中,每皿加9 ml 10%FBS+DMEM培养过夜。将已准备好的质粒与无血清DMEM、RNAi-Mate按比例混匀后转染293T细胞。37℃、5%CO2培养箱中培养8~10 h后,弃去上清液,再加入新鲜的10%FBS+DMEM,放入37℃、5%CO2培养箱中培养72 h。转染72 h后,将培养皿细胞上清液收集离心,浓缩液分装,-80℃保存备用。
1.2.4 病毒滴度测定 培养细胞融合至80%~90%时消化细胞,用培养液吹打成单细胞悬液。接种于96孔板中,混匀后培养24 h,然后用10倍稀释法将慢病毒稀释成3~5个梯度,每个孔加入100 μl稀释好的病毒液进行培养,24 h后弃去病毒液,加入含有10%血清的培养液继续培养72 h,通过荧光显微镜计数有荧光的细胞数,结合稀释倍数计算病毒滴度。病毒滴度(IU/ml)=(荧光细胞占总细胞数的百分比×接种细胞数/100×接种细胞体积)×1/稀释倍数。
1.3 子宫颈癌caski细胞的培养
将人子宫颈癌caski细胞置于含15%胎牛血清及1%青霉素(或链霉素)的RPMI 1640细胞培养基中,在37℃、5%CO2、80%湿度的细胞培养箱中传代培养。每天观察细胞生长状况。
1.4 病毒介导法将重组慢病毒感染caski细胞
caski细胞为HPV 16阳性的子宫颈癌细胞株;LV3-shNC为无目的基因的真核表达载体;LV3-shhTERT为HPV16 hTERT特异性siRNA表达载体。采用病毒介导法(逆转录病毒)将空载病毒LV3-shNC和携带LV3-shhTERT慢病毒分别以感染复数(MOI)为20和10感染子宫颈癌caski细胞,在37℃、5%CO2培养箱中感染8~10 h后,更换含有10%血清的完全培养基,继续培养3~5 d使病毒基因可整合到宿主细胞基因组,荧光显微镜下观察荧光数量以判断感染率。实验分为3组:空白对照组(未感染病毒的caski细胞)、阴性对照组(感染空载病毒LV3-shNC的caski细胞)、hTERT干扰组(感染携带LV3-shhTERT慢病毒的caski细胞)。
1.5 有效干扰序列的筛选
将转染72 h后的细胞消化至培养瓶中,继续培养24 h后,用已确定筛选浓度的嘌呤霉素培养液进行抗性筛选,每周换液2~3次,观察细胞生长情况。筛选14 d后将留下的抗性克隆团进行扩大培养,收集抗性克隆形成后1周的细胞。取部分细胞以流式细胞术仪检测荧光感染率,判断病毒的感染率。
1.6 qRT-PCR法检测hTERT mRNA的水平
常规法提取总RNA,逆转录为cDNA,qRT-PCR法检测hTERT mRNA的表达。hTERT基因引物:上游引物为5′-CTCCCATTTCATCAGCAAGTTT-3′,下游引物为5′-CTTGGCTTTCAGGATGGAGTAG-3′。GAPDH内对照引物:上游引物为5′-AGAAGGCTGGGGCTCATTTG-3′,下游引物为5′-AGGGGCCATCCACAGTCTTC-3’,引物均购自上海生工生物工程技术服务有限公司。扩增条件为95℃3 min,95℃12 s,62℃40 s,40个循环,25℃ 45 min退火。实验设3组,每项重复检测3次,取平均值。用2-△△Ct法计算各组目标基因mRNA模板的相对差异。△△Ct=[Ct(实验组目标基因)-Ct(实验组内参基因)]-[Ct(实验组目标基因)-Ct(实验组内参基因)],2-△△Ct=mRNA模板量的比值。
1.7 CCK-8法检测细胞活力
将3组实验细胞培养至对数生长期,细胞生长接近80%的融合度时,用0.25%胰酶消化,制成单个细胞悬液,细胞计数后在96孔板中每孔加入2 000个细胞,按照预实验的实验条件加培养液,每孔总体积为200 μl,共接种5个板,每块板都设空白组、NC-LV组、anti-htert-LV组,每组5个副孔,十字摇匀,放入细胞培养箱中培养24 h、48 h、72 h、96 h后每孔加入10 μl CCK-8溶液。继之在培养箱内孵育4 h,以酶标仪测定450 nm处的吸光度(OD值),以各组OD值计算细胞生存率,绘制细胞生长曲线。
1.8 统计学方法
采用SPSS 16.0统计学软件进行数据的统计分析。实验数据用均数±标准差(χ±s)表示,两组资料的比较用t检验;多组间的比较用单因素方差分析。以P<0.05为差异有统计学意义。
2 结果
2.1 重组hTERT质粒的鉴定结果
测序结果显示,针对hTERT基因的shRNA链与质粒pGLV3/H1/GFP+Puro连接成功。结果符合设计要求,表明实验成功构建了针对hTERT的shRNA质粒。见图1。

图1 慢病毒载体测序结果
2.2 慢病毒滴度测定结果
荧光显微镜观察显示,荧光细胞数目随病毒液的稀释倍数增加而逐步减少,按逐孔稀释滴度测定法测定其滴度为1×109TU/ml。见图2。
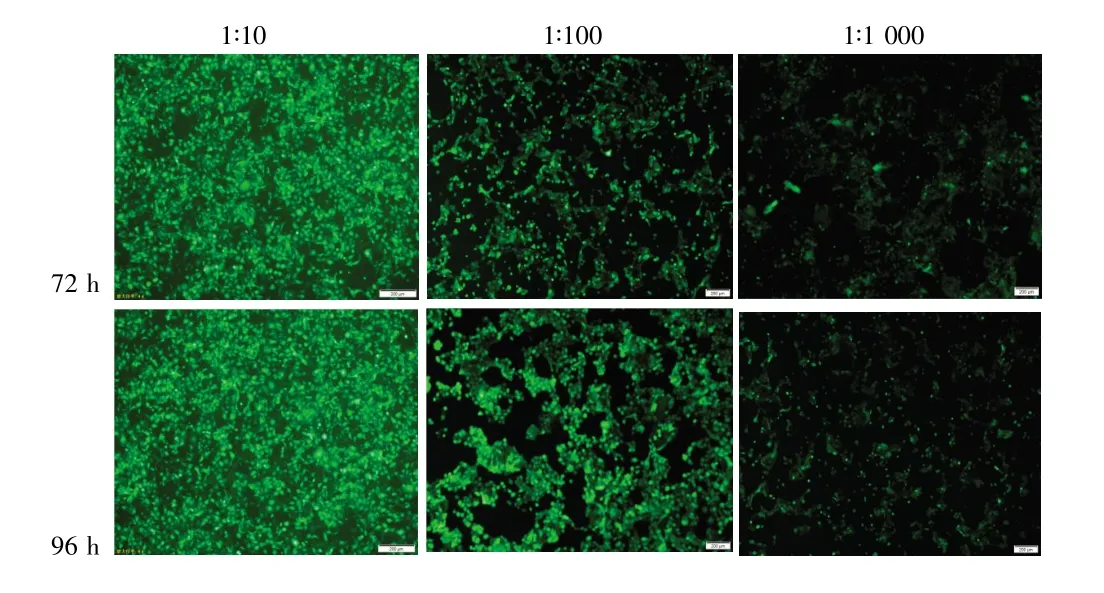
图2 不同稀释浓度的病毒原液侵染caski细胞的荧光表达(×40)
2.3 流式细胞术仪检测感染率
从目的细胞感染预实验测得caski细胞的MOI值为20,用一定浓度的嘌呤霉素筛选14 d后,经流式细胞术仪检测稳转细胞株荧光率为95%以上,达到本实验要求,可进行下游慢病毒介导的RNA干扰试验。见图3、图4。
2.4 实时荧光定量PCR检测hTERT mRNA的相对表达量
实时荧光定量PCR检测hTERT mRNA的相对表达量。结果转染后hTERT干扰组细胞中hTERT mRNA的表达量为0.97±0.053,明显低于空白对照组的1和阴性对照组的0.28±0.022(P<0.05);空白对照组和阴性对照组比较差异无统计学意义(t=1.6944,P>0.05)。见图5。
2.5 CCK-8法检测细胞增殖情况
CCK-8法检测结果显示:阴性对照组和hTERT干扰组感染后48 h、72 h、96 h,caski细胞的OD值差异有统计学意义(P均<0.05)。见表1。根据3组细胞OD值绘制细胞生长曲线,结果显示hTERT干扰组细胞生长速度较阴性对照组缓慢,细胞增殖受到抑制;而空白对照组和阴性对照组的细胞生长趋势基本一致。见图6。

图3 慢病毒感染caski细胞后用嘌呤霉素晒选后的荧光及常光照片(×40)

图4 流式细胞术仪分别测定慢病毒感染后的转染率及嘌呤霉素筛选后的感染率
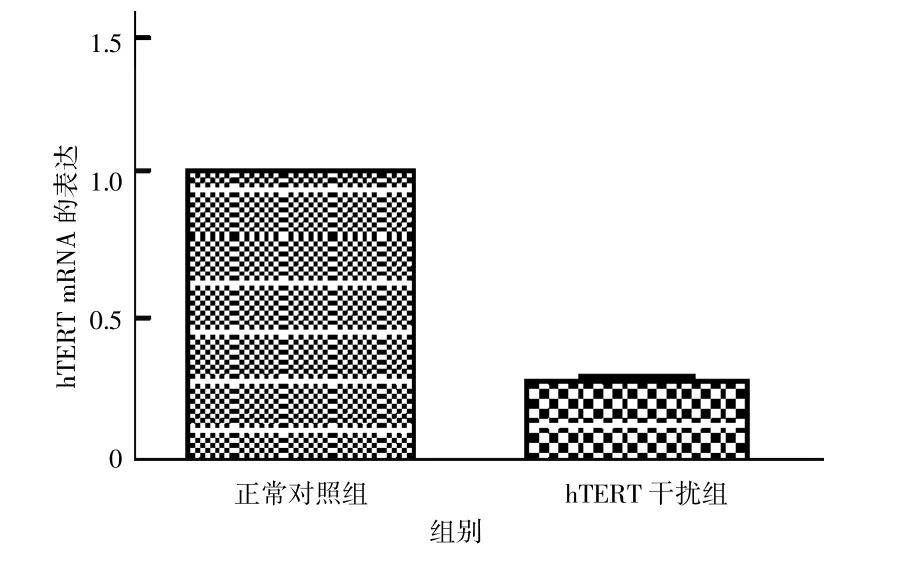
图5 感染慢病毒后hTERT mRNA的表达
表1 不同感染时间hTERT干扰组与阴性对照组子宫颈癌caski细胞OD值的比较(s)

表1 不同感染时间hTERT干扰组与阴性对照组子宫颈癌caski细胞OD值的比较(s)
感染后的时间(h)组别24 48 72 96阴性对照组 0.299±0.002 0.554±0.010 0.805±0.014 hTERT干扰组 0.301±0.008 0.356±0.007 0.396±0.013 0.475±0.008 t 0.408 10.317 19.076 41.610 P 0.697 <0.05 <0.001 <0.001 0.419±0.010

图6 慢病毒转染后各组子宫颈癌caski细胞的生长曲线
3 讨论
在恶性肿瘤的发生、发展过程中端粒酶具有重要意义,端粒酶RNA、相关蛋白和hTRET是组成人端粒酶的3种主要成分。临床数据表明,正常组织及大多数良性病变组织中端粒酶不表达或低表达,但90%以上的恶性肿瘤组织中端粒酶活性表达较高[8],而具有端粒酶活性的恶性肿瘤细胞均有hTERT的表达[9]。hTERT可能是特异性抑制肿瘤的良好靶点[10],当前以hTERT为靶点的RNA干扰基因沉默成为恶性肿瘤基因治疗研究的热点之一。有报道子宫颈癌组织中中可检测到大量hTERT的表达[11]。为进一步研究hTERT在子宫颈癌发生、发展过程中的作用,本实验将构建成功的hTERT-shRNA慢病毒表达载体感染子宫颈癌caski细胞,用CCK-8法检测hTERT-shRNA对caski细胞增殖活力的影响,结果发现hTERT干扰组感染后48 h、72 h、96 h细胞生长均较阴性对照组缓慢,提示细胞增殖受抑制,与莫小亮的报道结果一致[12]。
目前基因治疗已成为肿瘤治疗研究的焦点,而基因转染载体主要分为非病毒载体和病毒载体两大类。非病毒载体特异性差、转染率低且不能长久表达;病毒载体有反转录病毒、腺病毒和慢病毒等。研究发现,惟有慢病毒载体能高效感染分裂期和非分裂期细胞,并把基因高效整合到靶细胞,实现持久表达[13]。慢病毒属于逆转录病毒家族的一个亚类,能够携带目的基因高效整合到宿主细胞染色体中,不易引起突变,且不易诱发宿主免疫反应,能够稳定表达,具有较好的生物安全性,是一种理想的基因转移载体[14]。为了实现对子宫颈癌进行基因治疗[15],有研究人员曾应用核酶和反义核酸等技术干扰基因的表达,但结果发现特异性差、效率低且作用时间短暂,很难降解或敲除目的基因抑制[16,17]。RNAi是一种具有21Pb的序列特异的双链RNA(double-strandedRNA,dsRNA)在mRNA水平关闭相应序列基因的表达或使其沉默的过程. siRNA能高效导入细胞并且达到高效的基因沉默,其所需要的剂量远远小于核酶和反义核酸,具有高特异性、高效率、高成功率的特点,在肿瘤、病毒感染、遗传病及其他疾病的基因治疗中均有巨大潜力[18],已经开始应用于一些疾病临床治疗的研究[19~22],为特异性、个体化基因治疗以及肿瘤发病分子机制的研究提供新的技术手段[23]。有报道直接转染合成的siRNA虽能特异抑制哺乳动物细胞内同源基因的表达,有效抑制癌基因的表达[24],但细胞内siRNA很容易被降解[9],难以实现稳定的RNAi。为此,本研究采用慢病毒载体进行RNAi研究,与化学合成的siRNA和基于瞬时表达载体构建的普通siRNA载体相比,本研究构建的siRNA慢病毒载体不仅可以替代瞬时表达载体扩增使用,而且还可感染传统转染试剂难以转染的细胞系,如处于非分裂状态的细胞、悬浮细胞、原代细胞等,并整合入受感染细胞基因组内而实现稳定地长期表达,以便后续实验更为确切地观察结果奠定基础。
为成功构建hTERT-shRNA慢病毒表达载体,进一步阐明子宫颈癌发病与hTERT的关系,本研究利用RNAi技术设计和构建了针对靶基因hTERT的shRNA表达重组体,并经过第三代慢病毒pGLV3/H1/ GFP+Puro质粒载体包装、浓缩、收集病毒液,将构建成功的慢病毒载体感染子宫颈癌caski细胞,通过观测绿色荧光蛋白的表达判断转染成功与否以及估计转染效率,方法且简单、易行,干扰高效、特异性强且效果稳定。感染72~96 h后荧光显微镜下观查荧光表达及转染情况,嘌呤霉素筛选得到稳定的细胞株,流式细胞术仪测定转染率高达95%以上,达到实验要求,经实时荧光定量PCR检测细胞中hTERT mRNA的表达量明显降低。另外,本研究进一步用CCK-8法检测hTERT-shRNA对子宫颈癌caski细胞增殖能力的影响,沉默hTERT后的hTERT干扰组caski细胞生长速度缓慢。以上实验结果证明,本实验成功构建慢病毒表达载体,并建立anti-hTERT-LV稳转细胞系,有望为进一步深入探讨子宫颈癌发生、发展的分子机制提供新的特异性基因治疗靶点和技术,也为子宫颈癌的基因治疗应用于临床奠定实验基础。
[1] 吴令英,安菊生.宫颈癌治疗中的几个特殊问题[J].中国癌症防治杂志,2012,4(1):5-8.
[2] Corusi A,Skrgati L,Mahovli V,et al.Cervical cancer as a public health issue-what next?[J].Coll Antropol,2010,34(1):301-307.
[3] Howie HL,Katzenellenbogen RA,Galloway DA.Papillomavirus E6 proteins[J].Virology,2009,384(2):324-334.
[4] Wai LK.Telomeres.telomerase,and tumorigenesis-a review[J].Med Gen Med,2004,6(3):19.
[5] 贾海英,张涛,唐智,等.siRNA抑制喉鳞状细胞癌Hep-2细胞生长的作用[J].临床耳鼻咽喉头颈外科杂志,2010,24(7):308-310.
[6] Fire A,Xu S,Montgomery MK,et al.Potent and specific interference by double-stranded RNA in caenorhabditis elegans[J].Nature,1988,391(6669):806-811.
[7] Ui-Tei K,Naito Y,Takahashi F,et al.Guidelines for the selection of highly effective siRNA sequences for mammalian and chick RNA interference[J].Nucleic Acids Res,2004,32(3):936-948.
[8]Kirkpatrick KL,Mokbel K.The significance of human telomerase reverse transcriptase(hTERT)in cancer[J].Eur J Surg Oncol,2001,27(8):754-760.
[9] Wirth MG,Russell-Eggitt IM,Craig JE,et al.Aetiology of congenital and paediatric cataract in an Australian population[J].Br J Ophthalmol,2002,86(7):782-786.
[10]Reddy MA,Francis PJ,Berry V,et al.Molecular genetic basis of inheritedcataractandassociatedphenotypes[J].SurvOphthalmol,2004,49(3):300-315.
[11]Takakura M,Kyo S,Kanaya T,et al.Expression of human telomerase subunits correlation with telomerase activity in cervix cancer[J].Cancer Res,1998,58(7):1558-1561.
[12]莫小亮.HPV16E6和hTERT在宫颈癌致癌机制中的作用研究[D].南宁:广西医科大学,2014.
[13]Naldini L.Lentiviruses as gene transfer agents for delivery to non-dividing cells[J].Curr Opin Biotechnol,1998,9(5):457-463.
[14]尹巧云,李力,于红静,等.小分子蛋白慢病毒表达系统的构建[J].中国癌症防治杂志,2011,3(4):271-276.
[15]Alvarez-Salas LM,Ben í tez-Hess ML,DiPaolo JA.Advances in the development of ribozymes and antisense oligodeoxynucleotides as antiviral agents for human papillomaviruses[J].Antivir Ther,2003,8(4):265-278.
[16]Choo CK,Ling MT,Suen CK,et al.Retrovirus-mediated delivery of HPV16 E7 antisense RNA inhibited tumorigenicity of caski cells[J]. Gynecol Oncol,2000,78(3):293-301.
[17]Gottumukkala SN,Dwarakanath CD,Sudarsan S.Ribonucleic acid interference induced gene knockdown[J].J Indian Soc Periodontol,2013,17(4):417-422.
[18]Seyhan AA.RNAi:a potential new class of therapeutic for human genetic disease[J].Human Genetics,2011,130(5):583-605.
[19]Boudreau RL,Rodr í guez-Lebrón E,Davidson BL.RNAi medicine for the brain:progresses and challenges[J].Hum Mol Genet,2011,20(R1):21-27.
[20]Chen J,Xie J.Progress on RNAi-based molecular medicines[J].Int J Nanomedicine,2012,7:3971-3980.
[21]Yang WQ,Zhang Y.RNAi-mediatedgenesilencingincancertherapy[J]. Expert Opin Biol Ther,2012,12(11):1495-1504.
[22]Rao DD,Wang Z,Senzer N,et al.RNA interference and personalized cancer therapy[J].Discov Med,2013,15(81):101-110.
[23]Kubowicz P,Zelaszczyk D,Pekala E.RNAi in clinical studies[J].Curr Med Chem,2013,20(14):1801-1816.
[24]Verma NK,Dey CS.RNA-mediated gene silencing:mechanisms and its therapeutic applications[J].J Clinical Pharm Ther,2004,29(5):395-404.
[2014-10-14收稿][2014-11-26修回][编辑 罗惠予]
Construction of a plasmid expressing small hairpin RNA against human telomerase reverse transcriptase and functional analysis in the caski cervical cancer line
ZHAO Hong-ke△,MO Ling-zhao(Department of Gynecological Oncology,Affiliated Tumor Hospital of Guangxi Medical University;△Graduate School of Guangxi Medical University,Nanning 530021,P.R.China)
MO Ling-zhao.E-mail:molingzhao@hotmail.com
Objective To construct a plasmid expressing a small hairpin RNA(shRNA)to knockdown expression of human telomerase reverse transcriptase(hTERT),and to observe its effects on hTERT expression in caski cervical cancer cells.Methods Designer 3.0 software(Genepharma)was used to design an shRNA targeting hTERT.The shRNA was cloned into the pGLV3/Hi/GFP+Puro vector and confirmed by sequencing.Cells were divided into blank control group,negative control group and hTERT interference group.The resulting plasmid was transfected into caski cervical cancer cells and expression of the GFP reporter was analyzed.In addition,expression of chromosomal hTERT mRNA was quantified using RT-PCR.Cell proliferation in the presence and absence ofshRNA was measured using the cck-8 assay.Results A plasmid expressing an shRNA targeting hTERT was constructed and used to generate recombinant lentivirus.Lentiviral infection of caski cells led to lower expression of hTERT mRNA and slower cell proliferation than in controls.Conclusion A plasmid expressing shRNA targeting hTERT can effectively knockdown endogenous hTERT expression.This reagent may prove useful for understanding the role of telomerase in the pathogenesis of cervical cancer.
Cervical neoplasm;Human telomerase reverse transcriptase gene(hTERT);Lentivirus-based vectors;Small hairpin RNA;Caski cell
R737.33
A
1674-5671(2014)04-06
10.3969/j.issn.1674-5671.2014.04.05
广西自然科学基金资助项目(2013GXNSFAAO19254)
莫凌昭。E-mail:molingzhao@hotmail.com
猜你喜欢
——一道江苏高考题的奥秘解读和拓展
中学生物学(2022年7期)2022-09-07
成都医学院学报(2022年4期)2022-08-19
西南林业大学学报(2021年3期)2021-05-03
江西农业学报(2021年4期)2021-04-20
昆明医科大学学报(2021年1期)2021-02-07
康颐(2020年13期)2020-11-10
抗癌之窗(2020年2期)2020-07-14
抗癌之窗(2020年2期)2020-07-14
三农资讯半月报(2020年11期)2020-06-21
医药前沿(2018年22期)2018-01-17
