
真核生物启动子研究概述
2014-06-13 07:11李圣彦郎志宏黄大昉
生物技术进展 2014年3期
李圣彦, 郎志宏, 黄大昉
中国农业科学院生物技术研究所,农业部农业基因组学重点实验室(北京),北京100081
启动子是指RNA聚合酶识别、结合和启始转录的一段DNA序列,通常位于基因上游。启动子是调控基因表达的“指挥棒”,它能够控制基因表达的水平、部位及方式。深入研究启动子功能对于了解生物的生长发育、防御系统、疾病等都有非常重要的意义。本文总结一些研究启动子的方法及研究进展,以期能够为启动子的研究提供参考。
1 启动子的克隆
1.1 启动子捕获(promoter trap)技术
启动子捕获技术是一种克隆启动子的方法。该方法最早应用于大肠杆菌[1],后广泛应用于果蝇[2]、小鼠[3]和植物[4]等真核生物中。启动子捕获载体通常是一个包含报告基因的载体,在报告基因上游插入不同基因序列,如果可以驱动报告基因表达,则可判断插入片段具有启动子活性,进而从报告基因上游序列中分离出启动子序列(图1)[5]。
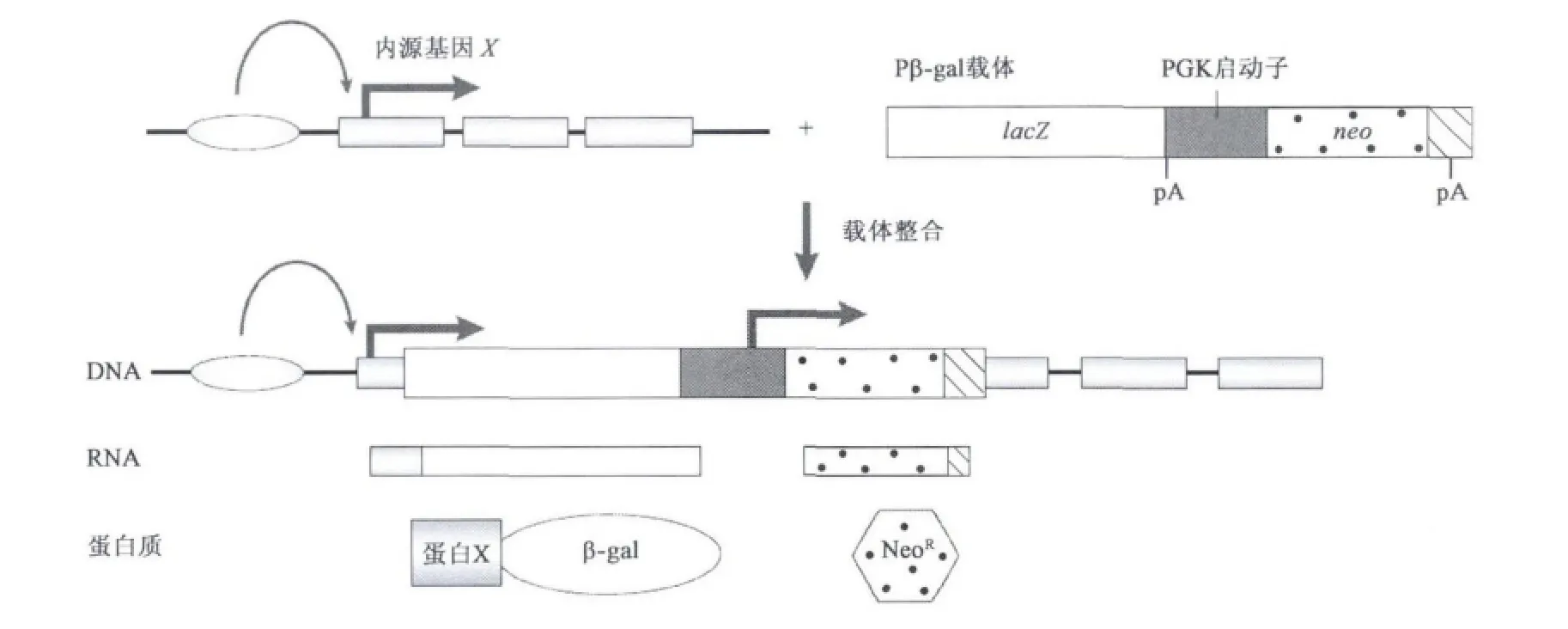
图1 启动子捕获载体示意图[6]Fig.1 A promoter trap vector diagram[6].
启动子捕获载体必须要正确的插入到外显子中才能表达,然而启动子捕获载体插入到外显子的频率非常的低[3,7],比增强子捕获载体的插入频率至少低200倍。因此启动子捕获载体通常含有筛选标记基因,如新霉素抗性基因(neo)或β-半乳糖苷酶-NeoR融合报告基因(图1),增加筛选的准确性。启动子捕获技术能够产生大量的突变,从中可以筛选出大量的启动子序列。但是这种插入位点的选择有一定的偏好性,因此该方法不能够在全基因组范围内产生近乎饱和的突变。德国人类基因组计划的基因捕获项目中利用电穿孔和逆转录病毒两种侵染方法将四种不同载体转入小鼠胚胎干细胞中,从获得的6 000个序列中捕获了4 587个基因[8,9]。他们的结果表明,利用多重载体和不同侵染方法可以最大限度的减小这种选择的偏好性。
1.2 基因芯片(gene chip)和RNA测序(RNA-seq)技术
基因的表达模式是由启动子控制的,那么了解了基因的表达模式就可以推测出其启动子的类型。由于技术手段的限制,之前多为研究一个或几个基因的表达模式,而对于某一条件下所有基因的变化情况无法了解。随着技术的发展,基因芯片(gene chip)和RNA测序(RNA-seq)技术使我们进行物种内某一条件下所有基因的表达量的研究成为可能。基因芯片,又称 DNA微阵列(DNA micro-array),是在一个芯片上固定大量已知序列探针,待测的靶标序列经过标记与芯片上特定位点的探针杂交,通过检测杂交信号,对生物细胞或组织中大量的基因信息进行分析。其突出特点在于高度并行性、多样性、微型化和自动化[10]。Schena 等[11]首次利用基因芯片技术检测不同拟南芥植株(野生型和转基因)及同一植株不同器官的45个基因的表达水平。通过与Northern杂交结果比对证明该技术可以实现自动化、高通量且准确地检测目的基因的表达水平。RNA-seq的研究对象为特定细胞或组织在某一功能状态下所能转录出来的所有RNA的总和,主要包括mRNA和非编码RNA。RNA-seq能够在全基因组范围内检测基因表达情况,进行差异基因筛选分析。该技术具有通量高、可重复性高、检测范围宽、定量准等特点,目前已被广泛应用。Lu等[12]利用RNA-seq技术全面分析籼稻和粳稻的转录组,鉴定出15 708个新的转录活性区(nTARs),其中51.7%与公开的蛋白数据无同源性,>63%推测为单外显子转录本。Zhang等[13]则利用该技术对栽培稻的8个器官的转录组进行了深度测序,发现约33%的水稻基因可发生可变顺式剪接,鉴定出234个推测的嵌合转录本。通过RNA-seq获得的大量新的转录本,对于我们发掘新的水稻启动子提供了很好的数据参考。对于研究基因的表达量而言基因芯片和RNA-seq这两种技术相比各有优缺点,基因芯片价格低廉,但是只能检测已知序列的信息而不能检测未知序列的信息;RNA-seq无需预先针对已知序列设计探针即可对任何生物整体转录活动进行检测,得到的数据也更加全面,但是价格比较昂贵。在研究中我们可以将两种方法结合起来使用,对于研究的物种先进行RNA-seq,利用RNA-seq得到的基因信息设计序列探针,然后利用基因芯片测定不同组织、不同条件下或不同处理后基因表达量的变化。现在许多基因芯片和RNA-seq结果都已共享,可以在相关数据库中下载到,例如GEO(http://www.ncbi.nlm.nih.gov/geo/)和 ArrayExpress(http://www.ebi.ac.uk/arrayexpress/)。通过分析已有的数据获得待选基因的表达量及表达模式,可以筛选出高表达及特异表达的基因,其相关的启动子序列即可通过PCR方法克隆获得。
2 启动子的生物信息学分析和预测
对获得的启动子序列首先利用生物信息学方法进行分析和预测,这将有利于我们了解启动子序列可能含有的一些功能元件,为下一步功能研究奠定基础。常用的植物启动子预测相关的数据库主要有:TRANSFAC、PLACE、PlantCARE、EPD及TRRD数据库等(表1)。
分析和预测植物启动子元件最常用的两个数据库是 PLACE和 PlantCARE。Hong等[19]及 Xu等[20]利用 PLACE数据库分别分析了拟南芥TPS11和TPS21基因以及棉花CAD1基因的启动子序列,发现拟南芥TPS11和TPS21基因的启动子序列中含有bHLH蛋白MYC2结合的E-box元件,通过实验证明MYC2能够直接与 TPS11和TPS21基因的启动子结合驱动这两个基因的表达,而在myc2突变体中,这两个基因的表达水平与野生型相比明显降低;分析棉花CAD1基因的启动子序列,发现其含有与 WRKY蛋白结合的G-box元件,在实验中从棉花中克隆10个WRKY基因,比较这10个基因在棉花品种 G.hirsutum L.cv Zhong-12(含棉酚)和 G.hirsutum L.cv GL-5(无棉酚)的表达情况,发现只有GaWRKY1在G.hirsutum L.cv Zhong-12中表达水平很高,其他9个基因没有明显差异或表达水平很低检测不到,推测GaWRKY1转录因子调控CAD1基因的表达。酵母单杂交试验则证明了GaWRKY1与CAD1基因启动子的G-box元件结合。在转基因拟南芥中过表达GaWRKY1基因也能够激活CAD1基因启动子连接的报告基因的表达,再次证明GaWRKY1转录因子能够调控CAD1基因的表达。上述研究均通过生物信息学分析获取基因启动子的重要功能元件信息,然后通过实验进行验证,发现了相应的转录因子等调控元件,对基因功能及调控的研究非常有益。
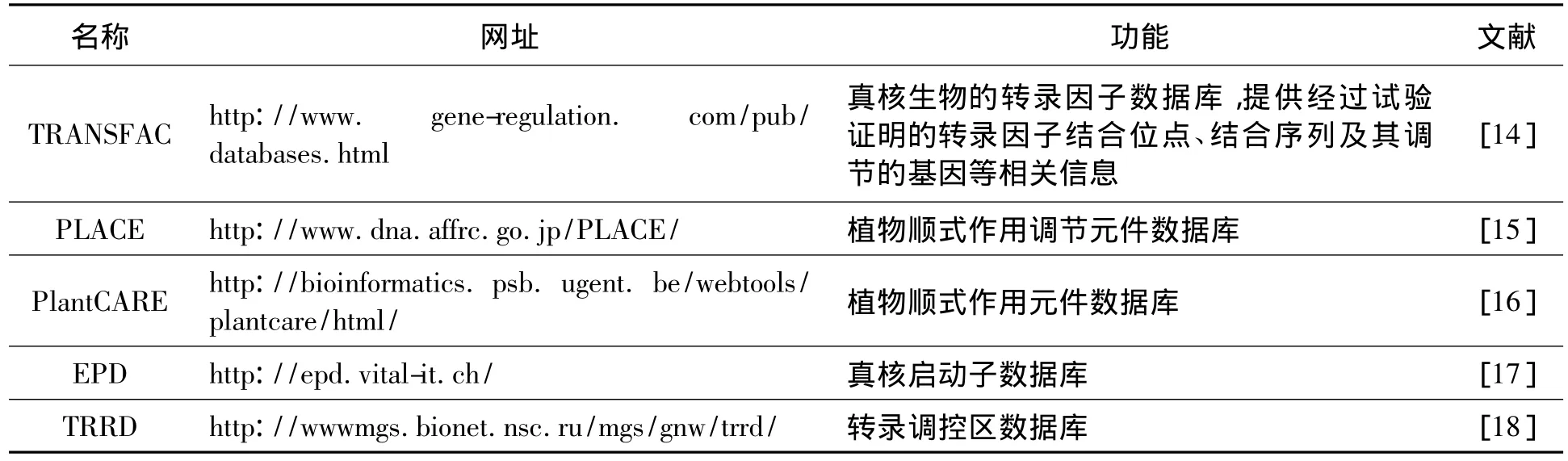
表1 植物启动子预测相关数据库Table 1 Database of plant promoter prediction.
3 启动子分析方法
3.1 片段缺失转化分析
利用生物信息学预测和分析启动子序列后,需要进行启动子缺失突变转化分析,目的是缩小启动子功能序列的范围。缺失突变主要有5'缺失、3'缺失、中间缺失及点突变,而常用的方法是构建一系列5'缺失的启动子片段连接报告基因的植物表达载体以确定启动子的功能序列的位置。目前所选择使用的报告基因主要有:β-葡萄糖苷酸酶基因(β-glucuronidase,GUS)、绿色荧光蛋白基因(green fluorescent protein,GFP)、氯霉素乙酰转移酶基因(chloramphenicol acetyl transferase,CAT)、荧光素酶基因(luciferase,Luc)和花青素基因(anthocyanin)等[21~23]。其中,GUS 和 GFP 报告基因因其操作方便、易于检测和定性而被广泛使用。Pan等[24]克隆了毛白杨4CL2基因的启动子,构建5'缺失启动子融合GUS报告基因表达载体转化烟草进行组织化学检测,发现当Pto4CL2启动子-317~-292 nt区域缺失后GUS基因不能在表皮和花瓣表达,而-266~-252 nt区域缺失导致组织特异性的丧失并且GUS活性也极大的降低。张文香等[25]从棉花中克隆了GhMADS29基因的启动子序列(1 316 bp)连接GUS报告基因构建植物表达载体转化拟南芥,组织化学染色分析发现其在14 d幼苗的根和叶中都有表达,在萼片、花瓣、雌蕊、果瓣中也表达,而在雄蕊和种子中不表达。推测GhMADS29可能与各种开花途径有关,与萼片、花瓣、雌蕊等花器官的发育有关,还可能与果实是否开裂有关。Brown等人[26]克隆了拟南芥PDF1.2基因的启动子序列(1 183 bp),构建了5'缺失启动子融合GUS报告基因的表达载体转化拟南芥,通过GUS活性检测发现缺失-261至-255的GCC box后转基因株系对茉莉酸的反应降低,这表明GCC box在PDF1.2启动子应对茉莉酸反应的过程中起着非常重要的作用。
转化材料的选择在启动子转化分析中非常重要。目前转化材料大多选用模式植物,如拟南芥和烟草。异源启动子在模式植物中分析的优点主要有:转化方法比较成熟,转化率较高;研究背景清晰,易于后续试验分析;生长周期较短等。缺点主要为由于种间差别,异源启动子在模式植物中表达模式并不能完全的模拟真实物种中的表达情况,并且对于一些物种特异的启动子可能在模式植物中并不表达或者表达模式迥异。
3.2 转录因子-启动子互作分析
与原核生物启动子相比,真核生物启动子要更为复杂,DNA的转录除了需要RNA聚合酶的作用外,还需要多种辅助因子。RNA聚合酶起始转录需要的辅助因子(蛋白质)称为转录因子(transcription factor,TF),它能够识别并结合DNA的顺式作用位点,从而调控目的基因以特定的强度并在特定的时间与空间表达。因此研究转录因子-启动子之间的相互作用关系,对于了解启动子的功能具有重要意义。传统研究蛋白质-DNA互作分析的方法主要有电泳迁移率变动分析(electrophoretic mobility shift assay,EMSA)、DNase I足迹法(DNase I footprinting)和甲基化干扰实验(Methylation interference assay)等,现在这些方法也在该领域中发挥着重要的作用。目前,应用较广的方法有酵母单杂交(yeast one-hybrid)实验及染色质免疫共沉淀法(chromatin immunoprecipitation,ChIP)。这两种方法出发点不同,酵母单杂交实验是利用已知的DNA序列筛选与该序列结合的蛋白质,而ChIP是利用已知蛋白分析该蛋白结合的DNA序列。
3.2.1 酵母单杂交 该技术可用来分析DNA与细胞内蛋白质相互作用,通过构建蛋白与转录激活区的融合表达载体,对酵母细胞内报告基因表达情况进行分析来筛选与DNA有特异结合的蛋白,该蛋白即对应一个基因文库中的编码核苷酸序列,因此广泛用于获得难于分离纯化的特定转录因子的蛋白质序列[27]。Spyropoulou 等[28]利用酵母单杂交技术以番茄SlTPS5基因启动子207 bp序列为靶标,从番茄cDNA文库中筛选出转录因子SlEOT1,该转录因子是一个类锌指蛋白,在番茄腺毛处表达,能够特异地转录激活SlTPS5基因。
但是该技术也存在一些缺陷。由于酵母自身蛋白可能与靶标DNA序列结合,或所用报告基因His或LacZ存在自泄露表达,在实际操作中常常出现漏检和假阳性现象。如果表达的融合蛋白对酵母有毒性、不能稳定存在或错误折叠等则会造成蛋白质不能与靶序列结合而产生假阴性现象。此外,酵母单杂交使用的cDNA文库是含有所有编码基因的文库,而编码转录因子的基因只占到很小的一部分,这也大大降低了酵母单杂交筛选的成功率。
为了提高酵母单杂交的成功率,科学家们研究出拟南芥的高效(或增强型)酵母单杂交系统[29~31]。该系统将拟南芥已知的转录因子克隆出来构建TF文库转入酵母Mata型菌株,诱饵载体转入酵母Matα型菌株,利用两种酵母交配型(Mata,Matα)能够接合实现酵母单杂交。该方法极大地提高了酵母单杂交的成功率,降低假阳性。但目前只见于拟南芥TF文库,其他植物物种的TF文库还未见报道。
3.2.2 染色质免疫共沉淀法 该方法简单而言就是利用甲醛等化学方法固定蛋白质-染色质复合体,然后通过超声波将染色质切割成大小适当的片段,通过特异抗体将目的蛋白-DNA片段的复合体免疫沉淀,获得的DNA片段可以克隆到适当的载体上经过测序即可得知目的蛋白结合的DNA 序列[32]。李玲等[33]利用 ChIP 证明转录因子RIN与LeACS2和LeACS4启动子区域的CArG-box序列结合。
随着测序技术和基因芯片的发展,在ChIP基础上发展出 ChIP-chip 和 ChIP-seq 技术[34~36]。但是由于DNA浓度及DNA片段的不均一性导致出现假阳性、漏检和mapping时位点不精确,为了提高精确度并降低 ChIP-chip的假阳性,Rhee等[37]又发展出ChIP-exo技术。该技术的主要原理为:蛋白质和DNA的复合物经过λ核酸外切酶(5'→3')处理后,裸露的DNA片段的5'序列会被降解至蛋白质结合位点处,3'序列保留做mapping,经过深度测序后与参考基因组比对,就能够得到蛋白结合DNA的准确位点,该方法能够精确到1个碱基。ChIP法检测灵敏,可直接显现DNA与蛋白质之间的体内动态结合,但难以同时得到多个转录因子对同一序列结合的信息。另外,如果研究的转录因子是间接的与启动子结合,也无法通过ChIP得到相关信息。
4 启动子的甲基化和多态性
除了转录因子结合到启动子上参与基因的表达调控外,启动子的甲基化(promoter methylation)和多态性(promoter polymorphism)都会影响基因的表达,因此对启动子甲基化和多态性的研究也非常广泛。
DNA甲基化是最早发现的表观遗传学修饰途径之一。DNA甲基化在真核生物和原核生物中均有发现,其对基因的表达具有非常重要的调控作用[38]。真核生物中DNA甲基化主要以5-甲基胞嘧啶的形式存在[39]。DNA甲基化通过改变染色质结构、DNA构象、DNA稳定性或者蛋白质与 DNA 相互作用方式[40,41],从而激活或抑制相关基因的表达。Zhang等[42]对拟南芥全基因组范围的DNA甲基化进行了检测,结果发现DNA甲基化主要存在于染色体的异染色质区域,如着丝粒;而在拟南芥已注释的30 334个基因中DNA甲基化更多的存在于假基因和非表达基因中,而不是在表达基因中。启动子甲基化调节基因表达的机制可能为启动子的甲基化区域能够与甲基化DNA结合蛋白相互结合,通过改变染色质的构象或者占据转录因子的结合位点从而阻断了转录因子与顺式元件的结合而导致转录的抑制[43]。
启动子多态性主要是指在基因组水平上由于单个核苷酸的变异所引起的启动子序列多态性。启动子多态性调控基因表达的原理可能为单个核苷酸的变异引起转录因子的结合位点的改变,从而影响基因的表达及表达方式。研究发现,人类的很多疾病都跟启动子的多态性有关系。Liu等[44]研究发现血管内皮细胞启动子-2578 C/A的多态性和-1154 G/A的多态性对于是否患有老年痴呆症有负相关效应。许多凋亡基因的启动子单核苷酸多态性(SNPs)会破坏细胞的程序性死亡过程,从而导致癌症的发生。FAS是一个转膜受体,属于肿瘤坏死因子(tumor necrosis factor,TNF)受体亚家族的一员,许多类型的细胞的凋亡信号传输都需要FAS的介导。Zhou等[45]研究发现FAS-1377G/A SNP能够改变FAS基因启动子的转录活性,在启动子-1377 G到A的转换破坏了刺激蛋白1(stimulatory protein 1,SP1)和信号转换激活转录蛋白1(signal transducer and activator of transcription 1,STAT1)的结合位点,衰弱启动子的活性进而降低FAS的表达,因此G能够保护细胞免受癌变而A可能是导致癌症的一个危险因子。
5 合成启动子
除了从自然界中发掘新的启动子外,还可以进行人工合成启动子。合成的启动子由核心启动子序列,主要的内含子,近端和远端的启动子序列组成,这些序列中可能含有特定的调节元件。合成的启动子与自然存在的启动子有非常大的不同,因为合成的启动子可以提供自然存在的启动子所没有的表达模式[46]。这种方法已经被成功的用于鉴定影响基因表达的调节元件和序列,并且由于合成的启动子所含元件都是根据需要安排进去的,所以合成的启动子能够做到非常准确的表达[47]。例如,DR5植物生长素启动子是一个高效的合成启动子,它含有串联的植物生长素响应元件TGTCTC,在植物中该启动子被用于研究植物生长素的响应机制[48]。
6 展望
启动子作为基因表达的重要调控元件,它的调控方式是多个层次多个因素共同作用的结果。近年来国内外对启动子的功能做了许多研究,也取得了很多进展。但由于现有的试验方法的局限性及启动子元件的复杂性,我们还不能完全了解启动子的功能。随着基因组学研究的不断深入和分子生物学研究方法的不断进步,会有更多更好的研究方法出现以解决目前启动子研究中存在的问题,推动启动子研究的发展。
[1]Casadaban M J,Cohen S N.Lactose genes fused to exogenous promoters in one step using a Mu-lac bacteriophage:in vivo probe for transcriptional control sequences[J].Proc.Natl.Acad.Sci.USA,1979,76(9):4530-4533.
[2]O'Kane C J,Gehring W J.Detection in situ of genomic regulatory elements in Drosophila [J].Proc.Natl.Acad.Sci.USA,1987,84(24):9123-9127.
[3]Friedrich G,Soriano P.Promoter traps in embryonic stem cells:a genetic screen to identify and mutate developmental genes in mice[J].Genes Dev.,1991,5(9):1513-1523.
[4]Chen S Y,Wang A M,Li W,et al..Establishing a gene trap system mediated by T-DNA(GUS)in rice.[J].J.Integr.Plant Biol.,2008,50(6):742-751.
[5]聂丽娜,夏兰琴,徐兆师,等.植物基因启动子的克隆及其功能研究进展[J].植物遗传资源学报,2008,9(3):385-391.
[6]Stanford W L,Cohn J B,Cordes S P.Gene-trap mutagenesis:past,present and beyond [J].Nat.Rev.Genet.,2001,2(10):756-768.
[7]Von Melchner H,DeGregori J,Rayburn H,et al..Selective disruption of genes expressed in totipotent embryonal stem cells[J].Genes Dev.,1992,6(6):919-927.
[8]de Angelis M H,Flaswinkel H,Fuchs H,et al..Genomewide,large-scale production ofmutantmice by ENU mutagenesis[J].Nat.Genet.,2000,25(4):444-447.
[9]Wiles M V,Vauti F,Otte J,et al..Establishment of a genetrap sequencetaglibrarytogeneratemutantmicefrom embryonic stem cells[J].Nat.Genet.,2000,24(1):13-14.
[10]滕晓坤,肖华胜.基因芯片与高通量DNA测序技术前景分析[J].中国科学 (C辑:生命科学),2008,38(10):891-899.
[11]Schena M,Shalon D,Davis R W,et al..Quantitative monitoring of gene expression patterns with a complementary DNA microarray[J].Science,1995,270(5235):467-470.
[12]Lu T,Lu G,Fan D,et al..Function annotation of the rice transcriptome at single-nucleotide resolution by RNA-seq [J].Genome Res.,2010,20(9):1238-1249.
[13]Zhang G,Guo G,Hu X,et al..Deep RNA sequencing at single base-pair resolution reveals high complexity of the rice transcriptome[J].Genome Res.,2010,20(5):646-654.
[14]Matys V,Fricke E,Geffers R,et al..TRANSFAC:transcriptional regulation,from patterns to profiles[J].Nucleic Acids Res.,2003,31(1):374-378.
[15]Higo K,Ugawa Y,Iwamoto M,et al..Plant cis-acting regulatory DNA elements(PLACE)database:1999[J].Nucleic Acids Res.,1999,27(1):297-300.
[16]Lescot M,Déhais P,Thijs G,et al..PlantCARE,a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences[J].Nucleic Acids Res.,2002,30(1):325-327.
[17]Périer R C,Junier T,Bucher P.The eukaryotic promoter database EPD [J].Nucleic Acids Res.,1998,26(1):353-357.
[18]Kolchanov N A,Ananko E A,Podkolodnaya O A,et al..Transcription regulatory regions database(TRRD):its status in 1999 [J].Nucleic Acids Res.,1999,27(1):303-306.
[19]Hong G J,Xue X Y,Mao Y B,et al..Arabidopsis MYC2 interacts with DELLA proteinsin regulating sesquiterpene synthase gene expression[J].Plant Cell,2012,24(6):2635-2648.
[20]Xu Y H,Wang J W,Wang S,et al..Characterization of GaWRKY1,a cotton transcription factor that regulates the sesquiterpene synthase gene(+)-δ-cadinene synthase-A [J].Plant Physiol.,2004,135(1):507-515.
[21]李 强.植物基因工程中常用的报告基因[J].生物学杂志,1995,(1):12-13.
[22]包满珠.植物花青素基因的克隆及应用[J].园艺学报,1997,24(3):279-284.
[23]薛丽香,童坦君,张宗玉.报告基因的选择及其研究趋势[J].生理科学进展,2002,33(4):364-366.
[24]Pan X,Li H,Wei H,et al..Analysis of the spatial and temporal expression pattern directed by the Populus tomentosa 4-coumarate:CoA ligase Pto4CL2 promoter in transgenic tobacco[J].Mol.Biol.Rep.,2013,40(3):2309-2317.
[25]张文香,范术丽,宋美珍,等.棉花 GhMADS29启动子克隆及表达分析[J].棉花学报,2013,25(4):309-315.
[26]Brown R L,Kazan K,McGrath K C,et al..A role for the GCC-box in jasmonate-mediated activation of the PDF1.2 gene of Arabidopsis [J].Plant Physiol.,2003,132(2):1020-1032.
[27]马洪波,杜 坚.酵母双杂交系统的研究进展与应用[J].中国国境卫生检疫杂志,2004,27(2):119-123.
[28]Spyropoulou E A,Haring M A,Schuurink R C.Expression of Terpenoids 1,a glandular trichome-specific transcription factor from tomato that activates the terpene synthase 5 promoter[J].Plant Mol.Biol.,2014,84(3):345-57.
[29]Mitsuda N,Ikeda M,Takada S,et al..Efficient yeast one-/two-hybrid screening using a library composed only of transcription factors in Arabidopsis thaliana [J].Plant Cell Physiol.,2010,51(12):2145-2151.
[30]Ou B,Yin K Q,Liu S N,et al..A high-throughput screening system for Arabidopsis transcription factors and its application to Med25-dependent transcriptional regulation [J].Mol.Plant,2011,4(3):546-555.
[31]Gaudinier A,Zhang L,Reece-Hoyes J S,et al..Enhanced Y1H assays for Arabidopsis [J].Nat.Methods,2011,8(12):1053-1055.
[32]Wells J,Farnham P J.Characterizing transcription factor bind-ing sites using formaldehyde crosslinking and immunoprecipitation[J].Methods,2002,26(1):48-56.
[33]李玲,傅达奇,朱毅,等.利用染色质免疫共沉淀技术确定转录因子 RIN调控的靶基因[J].生物技术通报,2011,(12):166-170.
[34]Ren B,Robert F,Wyrick J J,et al..Genome-wide location and function of DNA binding proteins[J].Science,2000,290(5500):2306-2309.
[35]Albert I,Mavrich T N,Tomsho L P,et al..Translational and rotational settings of H2A.Z nucleosomes across the Saccharomyces cerevisiae genome[J].Nature,2007,446(7135):572-576.
[36]Johnson D S,Mortazavi A,Myers R M,et al..Genome-wide mapping of in vivo protein-DNA interactions[J].Science,2007,316(5830):1497-1502.
[37]Rhee H S,Pugh B F.Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution [J].Cell,2011,147(6):1408-1419.
[38]Klose R J,Bird A P.Genomic DNA methylation:the mark and its mediators[J].Trends Biochem.Sci.,2006,31(2):89-97.
[39]Cao X,Jacobsen S E.Locus-specific control of asymmetric and CpNpG methylation by the DRM andcmT3 methyltransferase genes[J].Proc.Natl.Acad.Sci.USA,2002,99:16491-16498.
[40]Razin A.CpG methylation,chromatin structure and gene silencing—a three‐ way connection [J].EMBO J.,1998,17(17):4905-4908.
[41]Arnaud P,Goubely C,Pélissier T,et al..SINE retroposons can be used in vivo as nucleation centers for de novo methylation[J].Mol.Cell Biol.,2000,20(10):3434-3441.
[42]Zhang X,Yazaki J,Sundaresan A,et al..Genome-wide highresolution mapping and functional analysis of DNA methylation in Arabidopsis[J].Cell,2006,126(6):1189-1201.
[43]Curradi M, Izzo A, Badaracco G, et al.. Molecular mechanisms of gene silencing mediated by DNA methylation[J].Mol.Cell Biol.,2002,22(9):3157-3173.
[44]Liu S Y,Zeng F F,Chen Z W,et al..Vascular endothelial growth factor gene promoter polymorphisms and Alzheimer's disease risk:a meta-analysis[J].CNS Neurosci.Ther.,2013,19(7):469-476.
[45]Zhou Z X,Mi Y Y, Ma H Z, et al.. FAS-1377 G/A(rs2234767)polymorphism and cancer susceptibility:a metaanalysis of 17,858 cases and 24,311 controls[J].PloS ONE,2013,8(8):e73700.
[46]Hernandez-Garcia C M,Finer J J.Identification and validation of promoters and cis-acting regulatory elements[J].Plant Sci.,2014,(217-218):109-119.
[47]Rushton P J,Reinstädler A,Lipka V,et al..Synthetic plant promoters containing defined regulatory elements provide novel insights into pathogen-and wound-induced signaling[J].Plant Cell,2002,14(4):749-762.
[48]Ulmasov T,Murfett J,Hagen G,et al..Aux/IAA proteins repress expression of reporter genes containing natural and highly active synthetic auxin response elements [J].Plant Cell,1997,9(11):1963-1971.
猜你喜欢
中学生天地(A版)(2023年1期)2023-02-17
生命科学研究(2018年1期)2018-05-29
上海农业学报(2017年3期)2017-04-10
中国药理学通报(2015年2期)2016-01-12
热带农业科学(2015年9期)2015-10-14
中国医学科学院学报(2015年5期)2015-03-01
现代检验医学杂志(2015年2期)2015-02-06
沈阳医学院学报(2014年4期)2014-12-27
植物营养与肥料学报(2014年1期)2014-03-11
遗传(2014年3期)2014-02-28
