
食源性致病菌沙门氏菌与奇异变形杆菌二重PCR检测方法的建立*
2014-06-12 09:37张海霞孙桂珍孙振红
山东第一医科大学(山东省医学科学院)学报 2014年3期
张海霞 孙桂珍 王 慧 孙振红
(1.山东农业大学校医院;2.山东农业大学动物医学院,山东 泰安 271018 )
沙门氏菌和奇异变形杆菌是食品污染的常见致病菌,主要通过食品、饮水感染人类,导致细菌性食物中毒、肠热症、败血症等,甚至死亡。近年来,有少量文献报道奇异变形杆菌与沙门氏菌属具有共同抗原,能够与沙门氏菌属的多价血清和因子血清发生交叉凝集,引起菌种鉴定上的困难[1-2]。随着分子生物学检测的发展,尤其是在常规PCR的基础上发展起来的多重PCR检测技术,为实现多种致病菌的快速同时检测提供了广阔的发展空间[3]。因此本研究在常规PCR[4-6]检测奇异变形杆菌和沙门氏菌的基础上,通过优化PCR条件,建立其双重PCR检测体系,为快速同时检测奇异变形杆菌和沙门氏菌提供了新的有效手段。
1 材料与方法
1.1材料 菌株:奇异变形杆菌1株、沙门氏菌1株、普通变形杆菌1株、大肠杆菌1株、金黄色葡萄球菌1株、单核细胞增生性李斯特菌1株、蜡样芽孢杆菌1株,均由本实验室保存。主要试剂:rTaq、MgCl2、dNTP、10×PCR Buffer、Marker均购自北京全式金公司。引物:所用引物列于表1。以奇异变形杆菌的ureR基因和沙门氏菌的invA基因为研究对象,根据GenBank中基因序列,应用Primer 5.0引物设计软件设计两对特异性引物。引物均由南京金斯瑞科技有限公司合成。
1.2方法
1.2.1细菌的培养和模板的制备 分别挑取奇异变形杆菌和沙门氏菌两株标准菌单菌落接种于LB培养基,37℃振荡过夜培养。对菌落进行平板计数,配制108cfu/ml浓度两种细菌菌液,CTAB法[7]提取DNA。
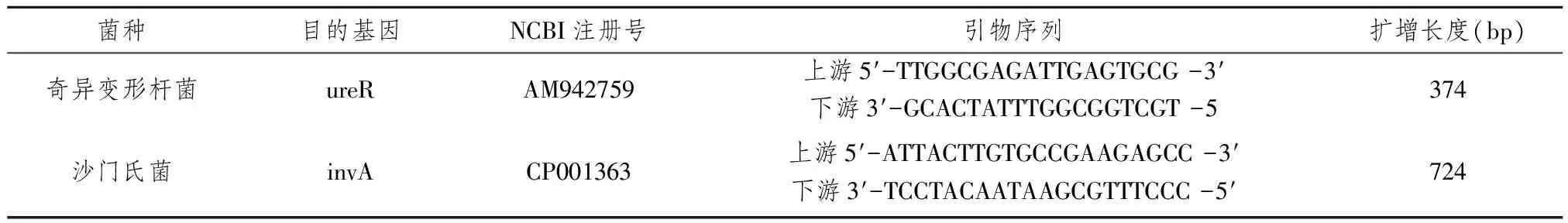
表1 引物序列及扩增产物
1.2.2单一PCR扩增及特异性和敏感性分析 先对2种菌进行单独扩增,反应体系为25 μl,分别为:10×Taq酶buffer 2.5 μl,MgCl2溶液2.0 μl,上、下游引物各1 μl(25 pmol/L),dNTP 2 μl(2.5 mmol/L),模板DNA 2 μl,Taq酶0.3 μl(5 U/μl), 其余用去离子水补足。PCR扩增条件:预变性95℃5 min;每个循环条件为94℃变性30 s,54℃退火30 s,72℃延伸50 s,30个循环;产物末端72℃延伸10 min。PCR扩增产物于1%的琼脂糖凝胶中,100 V电泳40 min后,观察产物条带。阳性克隆后送往南京金斯瑞科技有限公司进行核甘酸序列测定,与GenBank中发表的相应序列进行比较。
PCR特异性:奇异变形杆菌DNA模板分别加入ureR引物和invA引物,沙门氏菌DNA模板分别加入invA引物和ureR引物进行扩增。
PCR敏感性:分别将配制好的108cfu/ml浓度的两种细菌菌液,无菌操作按10倍递增稀释至浓度为108~101,CTAB法分别提取DNA,进行PCR检测,能检出的最低细菌浓度为该体系的灵敏度。
1.2.3单管多重PCR反应及反应体系的优化 模板DNA 4 μl(2种菌各2 μl),引物4 μl(2种引物各2 μl),dNTP 2 μl,Taq酶0.3 μl,10×Taq酶buffer 2 μl,MgCl2溶液3 μl,其余用去离子水补足,总体积25 μl,扩增条件同上。
多重PCR体系的优化采取固定其它因素,改变某一因素的方式,设计L16(43)正交试验,对影响多重PCR结果的扩增条件,如Mg2+浓度、引物浓度、dNTP浓度、退火温度等进行优化,以确定最佳的PCR反应条件。
1.2.4PCR特异性和敏感性测定 PCR特异性:两两混合1.1.1节中介绍的7种细菌,CTAB法分别提取DNA,在优化的多重PCR体系条件下扩增,进行特异性分析。PCR敏感性:将配制好的的108cfu/ml浓度的两种细菌菌液,无菌操作按10倍递增稀释,使菌液浓度为107~101cfu/ml。取各相同浓度菌液1 ml混合,后用CTAB法提取DNA,进行PCR检测后,计算检出限。
1.2.5人工模拟样品的检测 采用人工模拟污染经检测2种菌为阴性的肉馅样品,将2种菌的菌悬液10倍递增稀释至含菌量在10~1000 cfu/ml后,与肉馅混合,人工染菌的肉馅样品以1∶10加入相应的LB增菌液中,过夜培养后, CTAB法提取DNA,将以相同方法和时间制备的2种菌模板混合成为混合模板,进行PCR检测,同时对培养液进行培养计数。
2 结 果
2.1单一PCR扩增和核甘酸序列分析 奇异变形杆菌与沙门氏菌模板DNA在加入各自的引物扩增时,分别得到预计的PCR产物片段,即374 bp和724 bp。经克隆测序,奇异变形杆菌扩增片段与GenBank中AM942759 ureR基因序列同源性为98.9%,沙门氏菌扩增片段与GenBank中CP001363 invA基因序列同源性为99.6%。
2.2单一PCR特异性和敏感性分析 奇异变形杆菌模板DNA用invA引物扩增,沙门氏菌模板DNA用ureR引物扩增,均为阴性(图1)。PCR检测奇异变形杆菌的灵敏度达到了103cfu /ml(图2A),PCR检测沙门氏菌的灵敏度达到了104cfu /m l(图2B)。

M:DNA marker;1:ureR扩增奇异变形杆菌模板DNA;2:ureR扩增沙门菌模板DNA;3:invA扩增沙门菌模板DNA;4:invA扩增沙门菌模板DNA;5:阴性对照
图1 奇异变形杆菌、沙门氏菌单一PCR方法特异性检测结果

图2A 奇异变形杆菌单一PCR方法敏感性检测结果(M:DNA marker;1~8:奇异变形杆菌菌液浓度依次为108~101cfu/ml;9:阴性对照)
图2B 沙门菌单一PCR方法敏感性检测结果(M:DNA marker;1~8:沙门菌菌液浓度依次为108~101cfu/ml;9:阴性对照)
2.3单管多重PCR的扩增及其条件的优化 初步条件下在同一试管中可同时扩增2种目的基因片段,1%琼脂糖凝胶电泳显示在374 bp和724bp处有2条明显的条带存在。经试验优化得到如下的反应体系:模板DNA4μl(2种菌各2 μl)、上下游引物4μl(2种引物各2 μl)、dNTP 2.5 μl(2.5 mmol/L)、Taq酶0.5 μl(5 U/μl)、10×Taq酶buffer 2.5 μl、MgCl2溶液3 μl,其余用去离子水补足,总体积25 μl。PCR扩增条件同单重PCR。
2.4PCR特异性和敏感性测定结果 7种菌两两混合后,结果发现只有奇异变形杆菌和沙门氏菌出现特异性条带,而其它7种细菌均为阴性(图3)。从图4可以看出,在108~104cfu/ml浓度下,两菌均可同时扩增出较清晰条带(奇异变形杆菌在103cfu/ml浓度下仍然可见到条带),与单重PCR灵敏度一致,方法可取。

M:DNA marker;1:奇异变形杆菌和普通变形杆菌;2:沙门菌和大肠杆菌;3:奇异变形杆菌和沙门氏菌;4:金黄色葡萄球菌和大肠杆菌;5:单核细胞增生性李斯特菌和蜡样芽孢杆菌;6普通变形杆菌和大肠杆菌;7大肠杆菌和单核细胞增生性李斯特菌;8:大肠杆菌和蜡样芽孢杆菌;9:阴性对照
图3 奇异变形杆菌、沙门菌二重PCR方法特异性检测结果
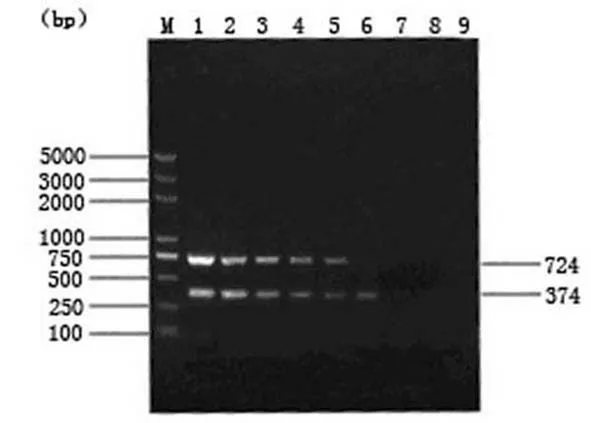
M:DNA marker;1~8:菌液浓度依次为108~101cfu/ml;9:阴性对照
图4 奇异变形杆菌、沙门菌二重PCR方法敏感性检测结果
2.5人工模拟结果 人工染菌肉馅中,染菌量在每克样品中奇异变形杆菌含量83 cfu,沙门菌含量40 cfu。经过增菌培养后,菌量分别达到奇异变形杆菌7.4×108cfu/ml,沙门菌4.4×107cfu/ml,运用前面的多重PCR体系进行检测,能够扩增出相应的特异性条带。实验证明人工染菌的样品能够同时检测出所接种的致病菌,并且无非特异性扩增,说明该多重PCR检测方法对食品中存在的奇异变形杆菌和沙门菌有很好的特异性和敏感性。
3 讨 论
多重PCR是在常规PCR基础上改进并发展起来的一种新型PCR扩增技术,可同时检测多种病原微生物,具有高效、高产、低成本、速度快等优点,目前已在多种病原微生物的检测中得到初步的应用[8-11]。本研究在参考别人研究的基础上,尝试用多重PCR检测奇异变形杆菌和沙门氏菌。
目的基因的筛选对PCR的特异性非常重要。奇异变形杆菌的ureR基因编码奇异变形杆菌脲酶调节子,林红乐等[12]通过对2株奇异变形杆菌、14株非奇异变形杆菌以及60份食物中毒送检标本进行检测,显示该基因具有很高的特异性。沙门氏菌的invA基因编码吸附和侵袭上皮细胞的表面抗原,李业鹏等[13]对77株沙门菌和24株非沙门菌进行检测,表明该基因也有很高的特异性。这都在本实验中得到了进一步的证实。同时,引物设计时已排除PCR扩增产物间可能发生的错配,避免引物对间形成黏性末端或部分双链,并对反应体系中各成分的使用量、比例、反应条件进行了优化,摸索出了最佳退火温度、引物浓度等,使实验达到了最佳效果。
本研究初步建立了快速检测志贺菌、沙门菌和霍乱弧菌的多重PCR体系,实现了对多种病原体DNA的同步快速扩增,为病原微生物的鉴定提供了快速、灵敏、特异的检测方法,具有较强的应用价值。进一步规范后可研制成检测试剂盒,从而为致病微生物检测试剂盒的推广应用奠定基础。
[1] 张元芳,钟小东,王顺东,等.三株与沙门菌有共同抗原的奇异变形杆菌的检出与分析[J].中国卫生检验杂志,2003,13:788-789.
[2] 王锦彤,钟广辉,熊定凯,等.珠江水中检出与沙门氏菌具有共同抗原的奇异变形杆菌[J].口岸卫生控制,2007,13(2):23-24.
[3] 张玉霞,黄鸣.食品检验中多重PCR技术的应用[J].中国卫生检验杂志,2008,18(5):958-960.
[4] Mansy MS, Fadl AA, Ashour MS, et al.Amplification of Proteus mirabilis chromosomal DNA using the polymerase chain reaction[J].Mol Cell Probes,1999,13(2):133-1401.
[5] Limanski A, Minukhin V, Limanskaia O, et al.Species-specific detection of Proteus vulgaris and Proteus mirabilis by the polymerase chainreaction[J].Zh Mikrobiol Epidemiol Immunobiol,2005,3:33-39.
[6] Charlotta Lfstrom, Rickard Knutsson, Charlotta Engdahl Axelsson, et al.Rapid and Specific Detection of Salmonella spp.in Animal Feed Samples by PCR after Culture Enrichment[J].Appl.Environ.Microbiol,2004,70:69-75.
[7] Ausubel FM, Brent R, Kingston RE, et al.精编分子生物学指南[M].颜子颖,王海林,译.北京:科学出版社,1998:39-40.
[8] 蔡亦红,姚余有.3种食源性致病菌的多重PCR快速检测方法的建立[J].中国卫生检验杂志,2007,17(11):1959-1962.
[9] 何超,樊学军.沙门菌、志贺菌和大肠杆菌O157:H7的多重PCR快速检测体系的初步探讨[J].卫生研究,2005,34(6):721-723.
[10] Mekonnen K, Divind E, Ruth-Anne S, et al.Amultiplex polymerase chain reaction assay for genus group and species-specific detection of mycobacteria[J].Diag Microbiol Infect Dis,2004,49:99-104.
[11] RYC Kong, SKY Lee.Rapid detection of six types of bacterial pathogens in marine waters by multiplex PCR[J].Water Res,2002,36:2802-2813.
[12] 林红乐,文岚,张如胜.PCR快速检测奇异变形杆菌的研究[J].实用预防医学,2009,16(2):560-562
[13] 李业鹏,钟凯.食品中沙门菌PCR检测方法的建立[J].中国食品卫生杂志,2006,18(1):17-22.
猜你喜欢
中国饲料(2022年5期)2022-04-26
中国土壤与肥料(2021年5期)2021-12-02
中国动物传染病学报(2021年3期)2021-07-21
疯狂英语·新悦读(2020年7期)2020-07-30
癌变·畸变·突变(2020年1期)2020-02-12
食品与机械(2019年1期)2019-03-30
中国卫生标准管理(2019年6期)2019-01-17
首都公共卫生(2018年5期)2018-11-21
上海蔬菜(2015年2期)2015-12-26
特产研究(2014年4期)2014-04-10
