
用随机森林算法预测六类酶的亚类*
2014-05-30 05:11王莹
阴山学刊(自然科学版) 2014年2期
王 莹
(齐齐哈尔工程学院,黑龙江齐齐哈尔 161000)
用随机森林算法预测六类酶的亚类*
王 莹
(齐齐哈尔工程学院,黑龙江齐齐哈尔 161000)
从蛋白质序列出发,我们首先将每条蛋白质序列分成等长的15段得到离散增量值、低频功率谱密度值、N端和C端的打分值和模体频数构成的组合向量表示蛋白质序列信息,用随机森林算法,对氧化还原酶、转移酶、水解酶、裂解酶、异构酶和连接酶中包含的亚类分别进行分类预测。总精度依次为90.86%、95.24%、96.42%、98.60%、97.53%和98.03%。除转移酶和水解酶略低于前人,其余好于前人的预测结果。
模体;矩阵打分值;离散增量;随机森林;酶的亚类
酶是具有高度专一性和催化效率的蛋白质,也称生物催化剂[1,2],完成生命过程中几乎所有的化学反应。由于酶的亚类决定其催化性及性能,对酶的亚类的研究非常必要。因此,本文对六类酶的亚类进行预测。从21世纪开始,对酶的分类出现了许多的预测方法,并取得了许多的研究成果。
2007年,Shen和Chou运用Top-down的方法预测酶的6个家族类和酶的亚类[3],其中酶的6个家族类的预测总精度为93.7%;各类酶的亚类预测总精度分别为 86.7%、95.8%、95.9%、94.4%、93.3%、98.3%。我们小组的石瑞佳等人采用支持向量机的方法预测酶的亚类得到较好的结果[4]。
由于随机森林算法是是处理高维、非线性模型的前沿理论和工具。因此,本文具体的采用随机森林(random forests,RF)算法构建一个非线性的模型,实现对六类酶的亚类进行分类预测。并得到较好的精度。
1 数据和方法
1.1 数据库
本文选取的数据集是通过ENZYME数据库http://www.expasy.org/enzyme/(released on 01 -May-2007)构建的新数据库,步骤如下:
(1)通过 ENZYME 和 Swiss- Prot(http://www.ebi.ac.uk/swissprot/)两个数据库得到酶的序列。
(2)首先去掉不足50个残基的序列。
(3)删除同时在两个或多个亚类中出现的序列,以免信息重复。
(4)利用BLAST软件使序列的相似性小于40%。
(5)为了保证信息的完整,最后将不足10条序列的亚类除去。
经通过处理后共得到14757条酶序列。其中氧化还原酶有2167条、转移酶有5460条水解酶有4498条、裂解酶的931条、异构酶有688条和连接酶有1013条。
1.2 参数选取
1.2.1 离散增量值(ID)

D(X+Y ) 为混合离散源
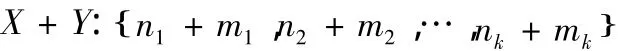
的离散量。计算公式为:

本文将一条酶序列分成15等份,分别用以紧邻疏水性的ID值作为参数。以氧化还原酶为例,对于一条酶序列由公式1可以得到18个离散增量值。其它各类作法类似。
1.2.2 低频功率谱密度值(F)
功率谱密度能反映序列的顺序信息,因此本文选取该值作为参数。提取功率谱密度值步骤如下[4]:
序列转换。利用氨基酸的疏水值(见表1,取自文献[4]),将酶序列转化成数字序列:R1R2…RL。其中Ri是第i个氨基酸。假设v(i)是第i个氨基酸残基的疏水值。
根据以下公式,对得到的数字序列进行离散傅里叶变换[9]:
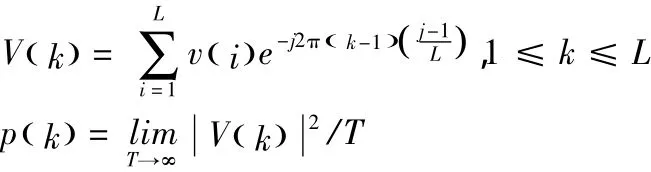
数据处理。通过以上的处理将每个序列都变成512维,表示512个频点。由于信号的特征及能量主要在低频部分[9],并通过多次验证得出取16时结果最好。因此,本文取每条序列前16个频点。又因为每条序列中第一个频点较特殊。因此,本文选取每条序列第2-16个低频功率谱密度值作为参数。
1.2.3 矩阵打分值(D)
矩阵打分算法[31-33]是一种有效的预测方法。我们利用该算法的打分函数来对酶序列进行特征提取。这里将矩阵打分算法分为以下3步介绍。
位置概率矩阵的构建。为更准确的表达序列信息,将氨基酸的位置概率作为位置概率矩阵的矩阵元[5]。
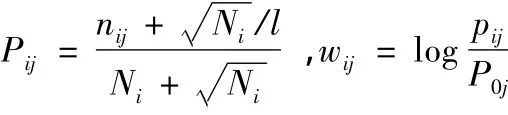
其中nij是第i个位置第j中氨基酸出现的频数,Ni是第i个位置出现氨基酸的总数,l为20。Pij表示第i个位置第j中氨基酸出现的概率。
位点保守性参量Ci反映位点氨基酸的保守性:
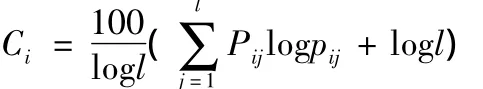
本文使用的打分函数为[13]:
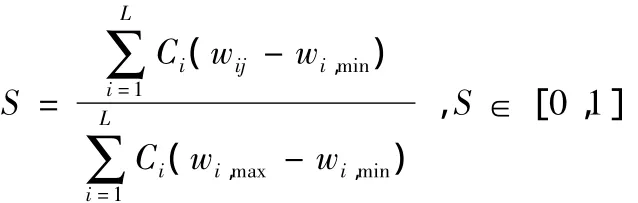
由于酶序列的N端和C端保守性比较好,因此本文选取N端和C端各30个氨基酸分别根据以上方法得到打分值。以氧化还原酶为例,对于一条蛋白质序列由公式2可以得到N端18个打分值和C端18个打分值。
1.2.4 模体频数值(MR)
模体通常与生物学功能相联系。为了反映序列功能信息。因此,本文利用MEME在线服务器(http://meme.nbcr.net/meme430/cgi- bin/meme.cgi[5])和 PROSITE 在线服务器(http://www.expasy.ch/prosite/[6])搜索各亚类的模体。
MEME。MEME中得到的模体具有统计学意义。因此选取每类亚类现次数最高的3种模体,以氧化还原酶的18个亚类为例,共得到54种模体。在一条酶序列中,对各模体进行计数。因此可以将序列变为频数值。
PROSITE。用Prosite数据库对酶序列进行搜索,得到N个模体。经过统计分析后,每类酶取三种出现较多的模体,以氧化还原酶的18个亚类为例,共得到18种模体。对各模体进行计数。因此可以将序列变为频数值。
1.3 随机森林算法(RF)
随机森林算法是2001年Leo Breiman提出的一种新型分类和预测模型[7-8],是基于决策树的分类器集成算法。它利用大数定律原理防止过度拟合、分类速度快,能有效处理大样本数据,能估计哪个特征参数在分类中更重要以及较强的抗噪声干扰能力等特点,因此,在基因芯片数据挖掘及药物筛选等生物学领域得到应用并取得了较好的效果。
本文随机森林算法是使用2.8.1版本的R软件(http://www.r-project.org/),通过调用其中的随机森林程序包来实现的。
2 结论
2.1 预测结果
将15段的六类亲疏水紧邻的离散增量值、低频功率谱密度、N端和C端氨基酸组分的矩阵打分值和两种模体频数值的组合向量共同输入到随机森林中,对六类酶的亚类分别进行预测,总精度依次为 90.86%、95.24%、96.42%、98.60%、97.53%和98.03%(见表1-6)。同时,我还将氧化还原酶个亚类与原库不同序列组成的数据集作为检验集,原数据库中的氧化还原酶作为训练集,进行独立检验,得到的预测总精度为96.11%。
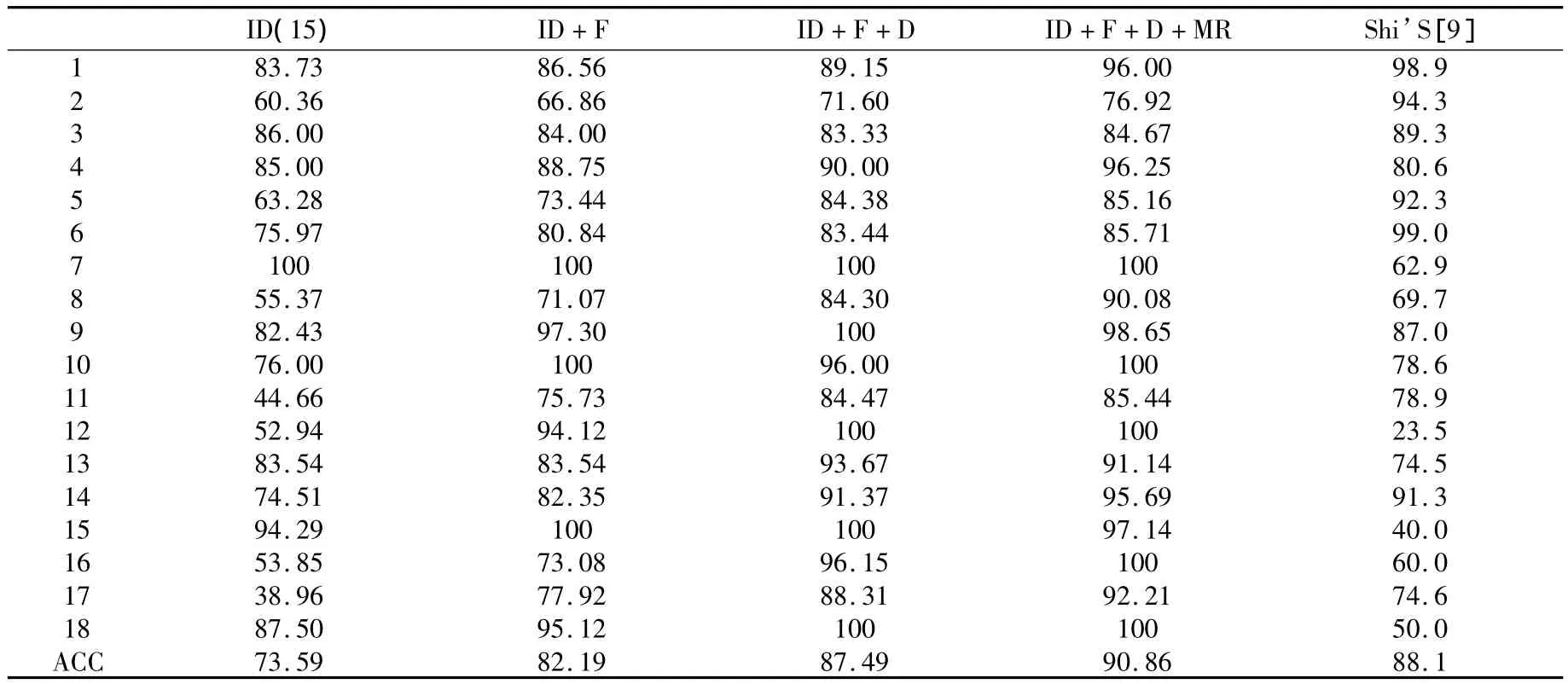
表1:氧化还原酶的18个亚类的结果
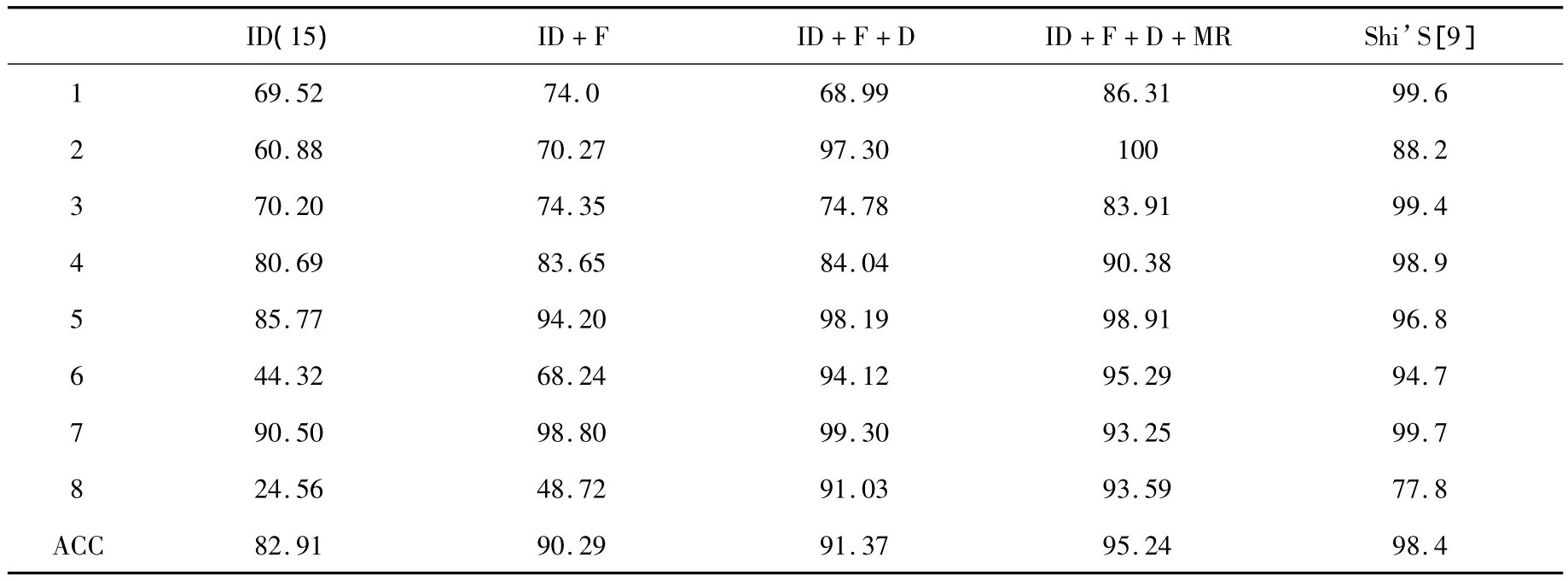
表2:转移酶的8个亚类的结果
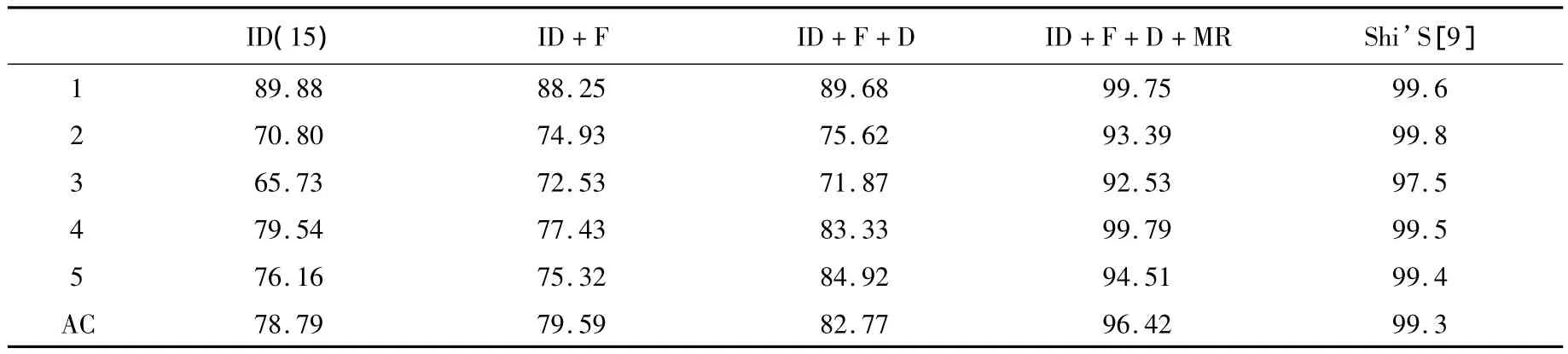
表3:水解酶的5个亚类结果
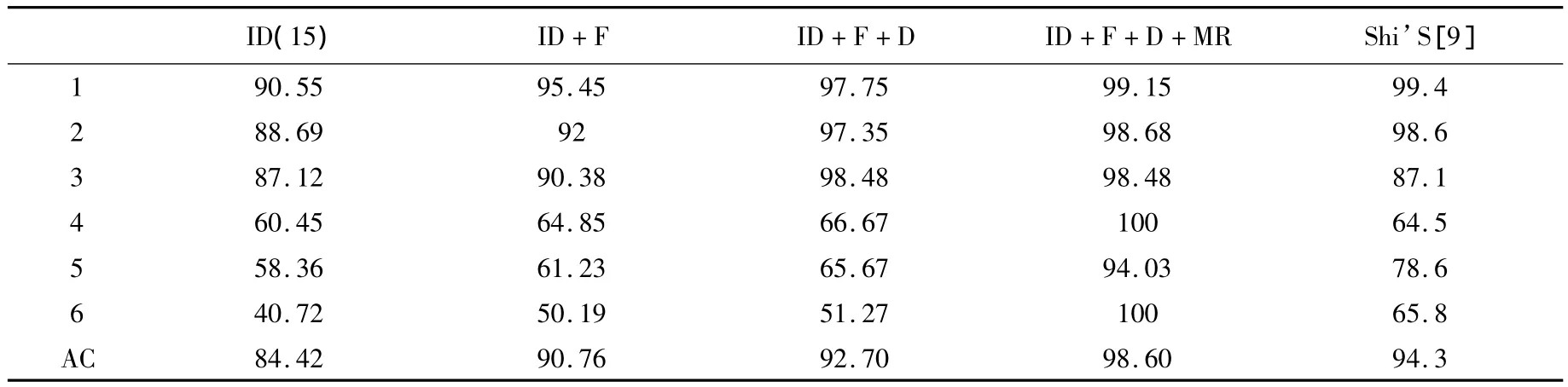
表4:裂解酶的6个亚类的结果
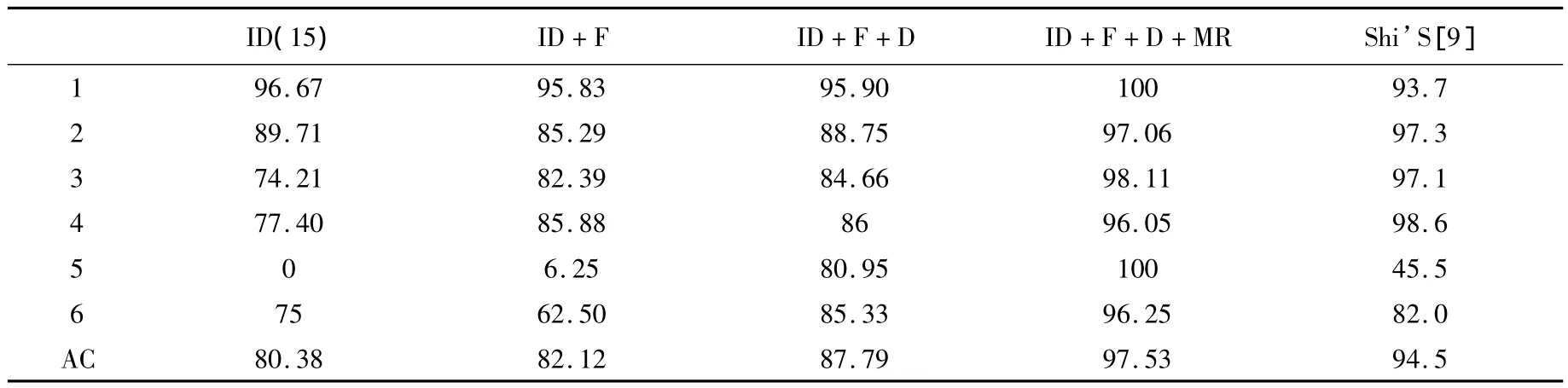
表5:异构酶的6个亚类的结果

表6:连接酶的6个亚类的结果
2.2 分析
根据上表,本文的预测方法是有效的,结果好于前人。其要原因:(1)随机森林算法是一种很有效的组合分类器,它由多棵决策树组成,最终的分类结果由所有决策树的综合投票而定,因此与单分类器(如SVM)算法相比分类更为准确。(2)利用模体反映序列功能,并使用了两种有效模体收索方法。(3)对序列进行分段处理,有效的突出了酶序列的活性部位,反映其有效信息。
[1]L.F.Yan,Z.R.Sun.Protein molecular structures[M].Beijing:Tsinghua University,1999:65–74.
[2]L.F.Yan.The structure and the function of protein[M].Changsha:Hunan science and technology publishing house,1988.
[3]SHEN H.B.,CHOU K.C.EzyPred:A top - down approach for predicting enzyme functional classes and subclasses[J].Biochemical and Biophysical Research Communication,2007,364:53-59.
[4]Ruijia Shi,Xiuzhen Xu.Predicting enzyme subclasses by using support vector machine with composite vectors[J].Volume 17,Number 5,May 2010:599-604(6).
[5]Bailey TL,Williams N,Misleh C,Li WW.MEME:discovering and analyzing DNA and protein sequence motifs[J].Nucl Acids Res,2006,34:369 -373.
[6]Castro,D.E,Sigrist,C.J.,Gattiker,A.,Bulliard,V.,etc.ScanProsite:detection of PROSITE signature matches and ProRule-associated functional and structural residues in protein[J].Nucleic Acids Research,2009,37:202 -208.
[7]Breiman L.Random forests[J].Machine Learning,2001,45:5-32.
[8]袁敏,胡秀珍.随机森林方法预测膜蛋白类型[J].生物物理学,2009,5:349-355.
[9]CHOU K.C.The biological functions of low - frequency phonons:3.Helical structures and microenvironment[J].Biophysical journal,1984,45:881 -890.
[10]Lei Liu,Xiuzhen Hu.Bases on Improved Parameters Predicting Protein Fold.2010 sixth international conference on Natural computation YanTai,shandon.
[11]李凤敏,李前忠.蛋白质亚细胞定位的识别[J].生物物理学报,2004,22(4):297-302.
[12]胡秀珍,李前忠.用离散量的方法识别蛋白质的超二级结构[J].生物物理学报,2006,22(6):424-428.
[13]杨科利,李前忠,林昊.预测酵母(Yeast)基因转录因子结合位点[J].内蒙古大学学报(自然科学版),2006,37(5):524-53.
Predicting Enzyme Subclasses by Using Random Forest
WANG Ying
(Qiqihar Institute of Technology;Qiqihar 161000)
Based on protein sequence,by selecting increment of diversity value,low - frequency of power spectral density,matrix scoring function values and motif frequency as characteristic parameters to describe the sequence information,Random Forest algorithm for predicting enzyme subclass is proposed.The overall success rate are 90.86%,95.24%,96.42%,98.60%,97.53%and 98.03%.Furthermore,in the same way,using the previous database to predict is better than the previous forecast results.
Motif;matrix scoring function value;Increment of diversity value;Random Forest algorithm;enzyme subclasses;prediction
Q55
A
1004-1869(2014)02-0022-04
10.13388/j.cnki.ysajs.2014.02.006
2014-04-17
王 莹(1986-),女,吉林永吉人,硕士,研究方向:生物信息学。
猜你喜欢
草业学报(2022年3期)2022-03-26
橡塑技术与装备(2022年3期)2022-03-17
昆明医科大学学报(2021年5期)2021-07-22
宁夏医学杂志(2020年3期)2021-01-21
智能计算机与应用(2019年1期)2019-01-11
中国实验诊断学(2017年5期)2017-06-05
自动化学报(2016年5期)2016-04-16
中华临床免疫和变态反应杂志(2014年2期)2014-04-08
中国酿造(2014年9期)2014-03-11
天津师范大学学报(自然科学版)(2014年4期)2014-02-18
