
巯基化阿霉素的两种合成方法的比较
2014-05-25 00:33吴珊,张叶叶,郭海霞等
药学实践杂志 2014年6期
·论著·
巯基化阿霉素的两种合成方法的比较
吴珊1,张叶叶1,郭海霞2,刘俊杰1,孙治国1,钟延强1,邹豪1(1.第二军医大学药学院药剂学教研室,上海 200433; 2.解放军266医院,河北承德 067000)
目的探索合成供金纳米粒载药系统研究用模型药物巯基化阿霉素的可行方法。方法 分别采用2-亚氨基硫烷盐酸盐(2-IT)法和琥珀酰亚胺-S-乙酰基硫代乙酸酯(SATA)法合成巯基阿霉素,通过高效液相色谱(HPLC)、飞行时间质谱(MS-ESI)及核磁共振氢谱(1H NMR)验证巯基阿霉素的合成,并考察反应物摩尔比、反应时间等因素对合成巯基阿霉素的影响。结果1 H NMR确证DOX-SATA出现了与硫酯基团相连的质子信号,表明新合成的化合物中含有硫酯基团。HPLC及MS-ESI结果显示,两种方法均能合成巯基阿霉素,2-IT法生成的巯基阿霉素,随着反应时间延长易发生环化,形成环化巯基阿霉素。SATA试剂法合成巯基阿霉素过程中不易发生副反应,合成的巯基阿霉素较为稳定。结论 通过两种方法的比较,SATA法合成巯基阿霉素的方法较为可行。
巯基化阿霉素;2-亚氨基硫烷盐酸盐(2-IT);琥珀酰亚胺-S-乙酰基硫代乙酸酯(SATA)
阿霉素(DOX)为放线菌(Streptomyces varcaesius)产生的蒽环类抗生素,是一种作用于DNA的药物,抗菌谱广,对多种肿瘤都有作用,广泛用于化学治疗。但长期使用阿霉素产生的蓄积性心脏毒性和肿瘤细胞对阿霉素产生的多药耐药性限制了阿霉素在临床上的应用[1]。
纳米载药系统具有增强肿瘤的EPR效应、减少游离药物的非特异毒性及延长药物的体循环时间,增强药物在作用部位的蓄积,提高药效等优点而成为逆转肿瘤多药耐药的有效逆转策略之一[2]。近年来基于阿霉素的纳米载药系统主要包括阿霉素脂质体[3]、阿霉素胶束[4]、阿霉素聚合物纳米粒[5]等,其中,以Au-S键为基础的谷胱甘肽还原响应型纳米载药系统[6]成为研究热点。
为使阿霉素能与金纳米粒通过Au-S键形成金纳米粒载药系统,首先需要对游离的阿霉素进行化学修饰,合成具有含有游离巯基的巯基阿霉素(DOX-SH)。本研究采用两种不同方法合成了模型药物DOX-SH,并通过高效液相色谱(HPLC)、飞行时间质谱(MS-ESI)及核磁共振氢谱(1H NMR)比较了这两种方法的可行性,为进一步构建金纳米粒载药系统奠定基础。
1 材料与仪器
1.1 材料盐酸阿霉素(DOX·HCl)(大连美仑生物技术有限公司);2-亚氨基硫烷盐酸盐(2-IT)、琥珀酰亚胺-S-乙酰基硫代乙酸酯(SATA)(美国Sigma公司);N,N-二甲基甲酰胺(DMF)、三乙胺(TEA)、乙酸乙酯(Ethyl acetate)、氯化钠(NaCl)、盐酸羟胺(NH2OH·HCl)、碳酸钠(Na2CO3)(上海国药集团化学试剂有限公司);三氟乙酸(TFA)(美国Tedia公司);乙腈、甲醇均为色谱纯(德国默克公司)。实验用水均为去离子水。
1.2 仪器85-2恒温磁力搅拌器(中国国华电器有限公司),Nanopure DiamondTM纯水机(美国Barnstead公司),AL104电子天平[METTLER TOLEDO仪器(上海)有限公司],分液漏斗(上海和器公司),浓缩仪(德国Eppendorf公司),高效液相色谱仪LC-20A(日本岛津公司SHIMADZU),十万分之一天平[METTLER TOLEDO仪器(上海)有限公司],超声波清洗器SK 3300LH(上海市科导超声仪器有限公司),核磁共振仪Avane II 600 MHz(瑞士Bruker公司),飞行时间质谱仪G6220A TOF(美国Agilent Technologies)。
2 方法
2.1 2-IT试剂法合成DOX-SH[7]
2.1.1 制备方法精密称取DOX·HCl和2-IT按1∶10、1∶20、1∶30的摩尔比混合后溶于适量甲醇中,加入适量三乙胺,调节反应体系的pH值为8~10,在N2保护避光的条件下,室温搅拌2 h或24 h后,纯化干燥得到DOX-SH。
将反应后的产物进行HPLC色谱分析,考察不同反应物摩尔比及不同反应时间对DOX-SH合成的影响。
2.1.2 HPLC检测DOX-SH色谱条件色谱柱: ODS色谱柱;柱温:30°C;流动相:乙腈和水(0.2% TFA)(35∶65,V/V);流速:1.0 ml/min;紫外检测波长:490 nm;进样量:20μl;保留时间:20 min。
2.1.3 不同反应摩尔比对DOX-SH的合成影响反应摩尔比是影响反应进程的关键因素之一,本研究考察DOX·HCl与2-IT摩尔比分别为1∶10、1∶20、1∶30对合成DOX-SH的影响,反应时间为2 h,计算不同反应摩尔比阿霉素的转化率。
2.1.4 不同反应时间对DOX-SH的合成影响根据“2.1.3”确定的最优反应摩尔比按照“2.1.1”的反应流程进行反应,分别在2 h和24 h对产物进行HPLC色谱分析,考察不同反应时间对DOX-SH合成的影响。
2.2 SATA试剂法合成DOX-SH
2.2.1 制备方法[8]精密称取10.5mg DOX·HCl和25.2mg SATA于10ml试管中,用3ml DMF溶解,加入6μl三乙胺于磁力搅拌器上搅拌3 h得到乙酰硫代乙酸酯取代的阿霉素(DOX-SATA),反应后的溶液用5ml水稀释,分别用8、7ml乙酸乙酯进行萃取,合并乙酸乙酯层,依次用1%盐酸、饱和碳酸钠、饱和食盐水洗涤3次,旋转蒸发浓缩得到第一步反应产物DOX-SATA。取纯化后的DOX-SATA适量溶于DMF中,加入3.4mg 0.5mol/L盐酸羟胺、2μl三乙胺继续反应1 h后,纯化干燥得到DOX-SH。
2.2.2 HPLC验证不同时间各步反应产物及产物稳定性取“2.2.1”中反应生成的第一步产物DOX-SATA及终产物DOX-SH分别溶于流动相后,HPLC分析各步产物。将制备的DOX-SATA和DOX-SH室温放置24 h后,HPLC分析各产物的稳定性。
2.3 飞行时间质谱MS-ESI验证DOX-SH的合成将“2.1”及“2.2”中制得的DOX-SH分别溶于甲醇,于飞行时间质谱仪G6220A TOF进行测定,检测两种方法合成的DOX-SH的分子量。
质谱条件:检测模式为负离子模式;雾化器压力0.34 MPa;毛细管电压3 600 V;碎片器电压120 V;氮气流量10 L/min;氮气温度300.0°C。
2.4 核磁共振氢谱(1H NMR)验证DOX-SATA的合成称取10 mg DOX、DOX-SATA粉末,分别溶于MeOD-d4,用600 MHz核磁共振仪(AvaneⅡ600 MHz,Bruker)鉴定化合物的分子结构。
3 结果
3.1 2-IT试剂法合成DOX-SH2-IT试剂法合成DOX-SH的反应机制如图1所示,2-IT为伯胺的硫醇化试剂,DOX上的伯胺与2-IT试剂反应初期快速形成含有游离巯基和不稳定的亚氨基的DOX-SH,但是DOX-SH在pH值为7.8的条件下,分子内部的亚氨基极易与游离巯基的氢原子结合脱去氨气,形成环化巯基阿霉素(cyc-DOX)。
3.1.1 不同反应摩尔比对合成DOX-SH的影响用2-IT试剂法合成DOX-SH,其合成的转化率受到反应物的摩尔比的影响。图2A中DOX的保留时间为7.1 min,图2B~2D中3份色谱图均出现了保留时间分别为12.7 min的DOX-SH和10.6 min的cyc-DOX的色谱峰。由图可知,增大反应摩尔比,DOX-SH和cyc-DOX的色谱峰峰面积增大,表明增大反应产物的摩尔比,DOX-SH的产率增加。图2D表示,DOX与2-IT试剂反应2 h,DOX基本反应完全。以DOX的浓度为横坐标,色谱峰面积值为纵坐标进行线性回归得到标准曲线方程为:A= 14 587C-15 620(r=0.999 6)。根据标准曲线计算得到不同反应摩尔比DOX的转化率如表1所示。
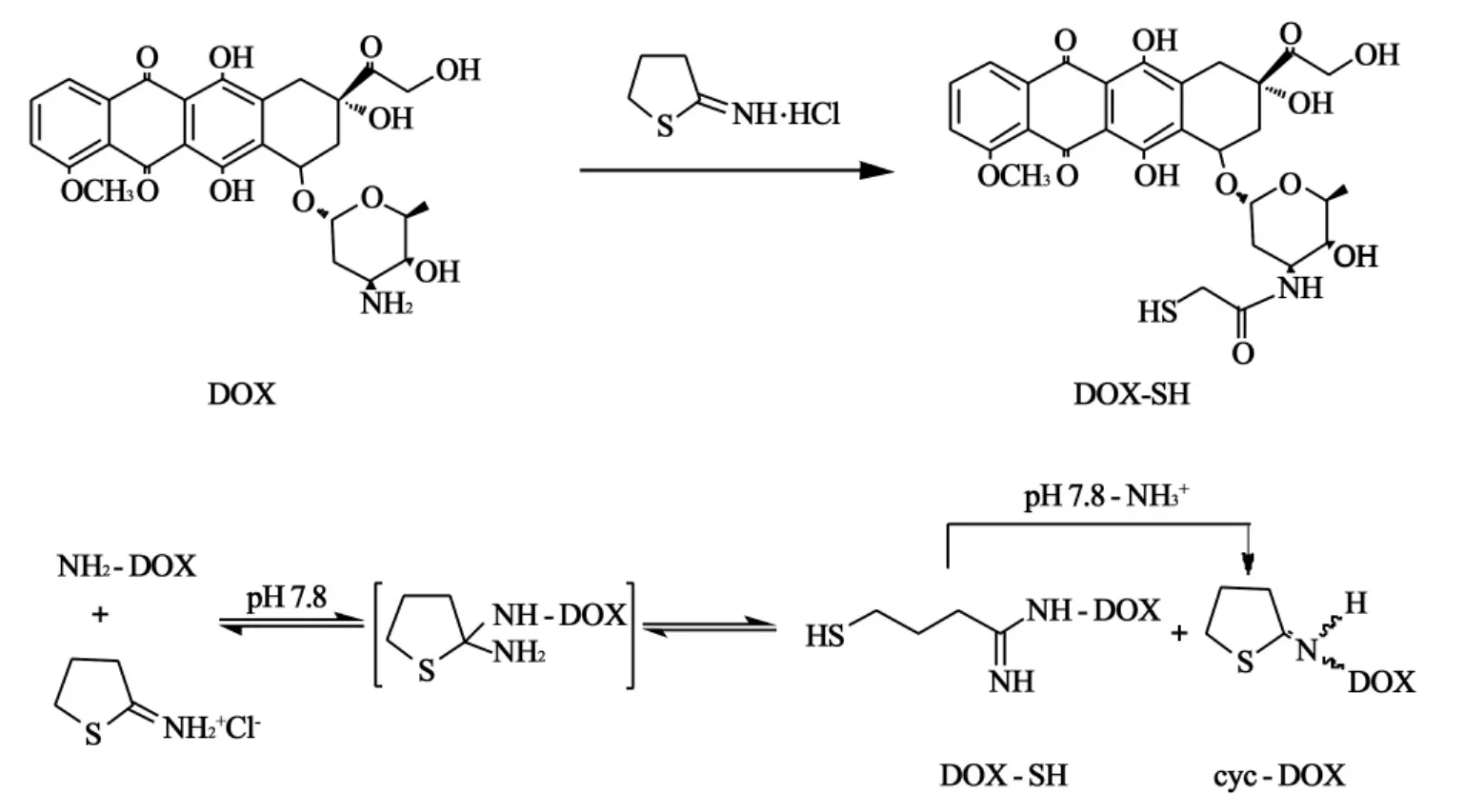
图1 2-IT试剂法合成DOX-SH反应机制
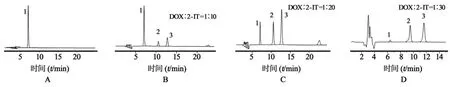
图2 不同反应摩尔比合成DOX-SH的HPLC色谱图

表1 2-IT试剂法合成DOX-SH不同反应摩尔比DOX的转化率
3.1.2 不同反应时间对合成DOX-SH的影响本研究进一步利用HPLC法考察了不同反应时间对DOX-SH合成的影响。图3A、图3B为DOX与2-IT试剂反应2 h和24 h后的DOX-SH色谱图,图3B与图3A相比,DOX的色谱峰峰面积减小,甚至消失,表明反应时间延长,DOX反应完全。但是与此同时,DOX-SH色谱峰也消失,表明该DOX-SH不稳定,cyc-DOX明显增加。

图3 不同反应时间合成DOX-SH的HPLC色谱图A(2 h)、B(24 h)
3.2 SATA试剂法合成DOX-SH
3.2.1 HPLC色谱分析合成DOX-SH各步反应产物合成DOX-SH也可采用双异官能团交联剂SATA试剂法,该方法合成DOX-SH的反应机制如图4所示。琥珀酰亚胺-S-乙酰基硫代乙酸酯SATA与伯胺反应添加保护性巯基。DOX的伯胺和SATA上琥珀酰亚胺在一定摩尔比下反应生成中间产物DOXSATA,DOX-SATA的乙酰基硫代乙酸酯在盐酸羟胺的作用下脱去乙酰基团,最终在DOX分子上引入游离巯基基团。
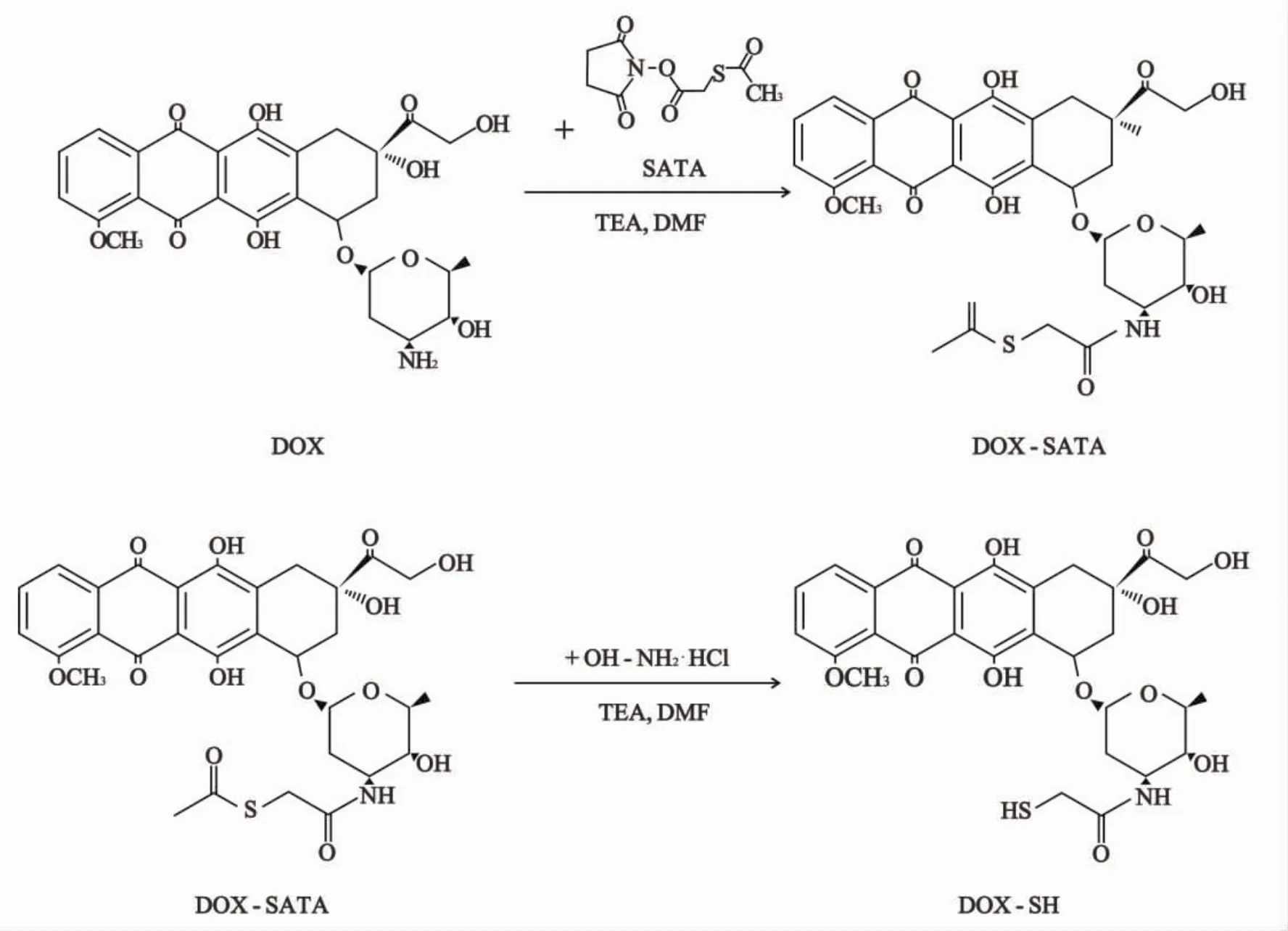
图4 SATA试剂法合成DOX-SH的反应流程图
将该方法合成DOX-SH过程中的各部分产物按照“2.1.2”项下色谱条件进行色谱分析。图5A保留时间为6.4 min,反应3 h后生成的DOX-SATA的保留时间为14.8 min(图5B),经盐酸羟胺脱去乙酰基团后生成DOX-SH的色谱图如图5C所示,保留时间改变为9.9 min。

图5 SATA试剂法合成DOX-SH的各步反应产物的HPLC色谱图
3.2.2 HPLC验证DOX-SATA及DOX-SH的稳定性
SATA试剂法合成DOX-SH各步反应产物的稳定性如图6所示,从图6A可以看出,放置24 h后 DOX-SATA的保留时间(15.26 min)变化不大,表明DOX-SATA比较稳定。与反应初生成的DOX-SH相比,也并未发生明显变化,表明该方法合成的DOX-SH较为稳定(图6B)。
3.3 MS-ESI验证两种方法DOX-SH的合成通过MS-ESI考察了两种合成DOX-SH的分子量,图7A为未发生反应的DOX的质谱图,阿霉素的分子离子峰为544.18,经2-IT试剂法巯基化之后的DOX-SH的分子离子峰为645.21(图7B),分子量为628.18的分子离子峰与化合物的相对摩尔质量645.21相比,分子量减少了17,结合“3.1.1”中2-IT试剂与阿霉素反应的机制可知,628.18为cyc-DOX的分子离子峰,该结果与HPLC色谱图结果一致。SATA试剂法合成的DOX-SH如图7C所示,分子离子峰为616.15,两种方法合成的DOXSH的分子量均与理论值相符,表明两法均能成功合成DOX-SH。
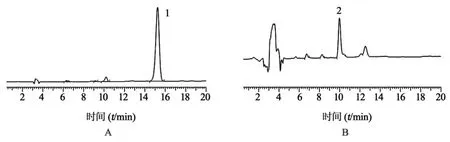
图6 SATA试剂法合成DOX-SH各步反应产物放置24 h后的稳定性1.DOX-SATA;2.DOX-SH

图7 DOX(A)、DOX-SH(B,2-IT试剂法)、DOX-SH(C,SATA试剂法)的MS-ESI谱图
3.41H NMR验证DOX-SATA的合成采用600 M1H NMR和HPLC法对DOX的巯基化反应产物进行鉴定。如图8所示,与DOX相比,DOX-SATA出现了与硫酯基团相连的质子信号,表明新合成的化合物中含有硫酯基团。
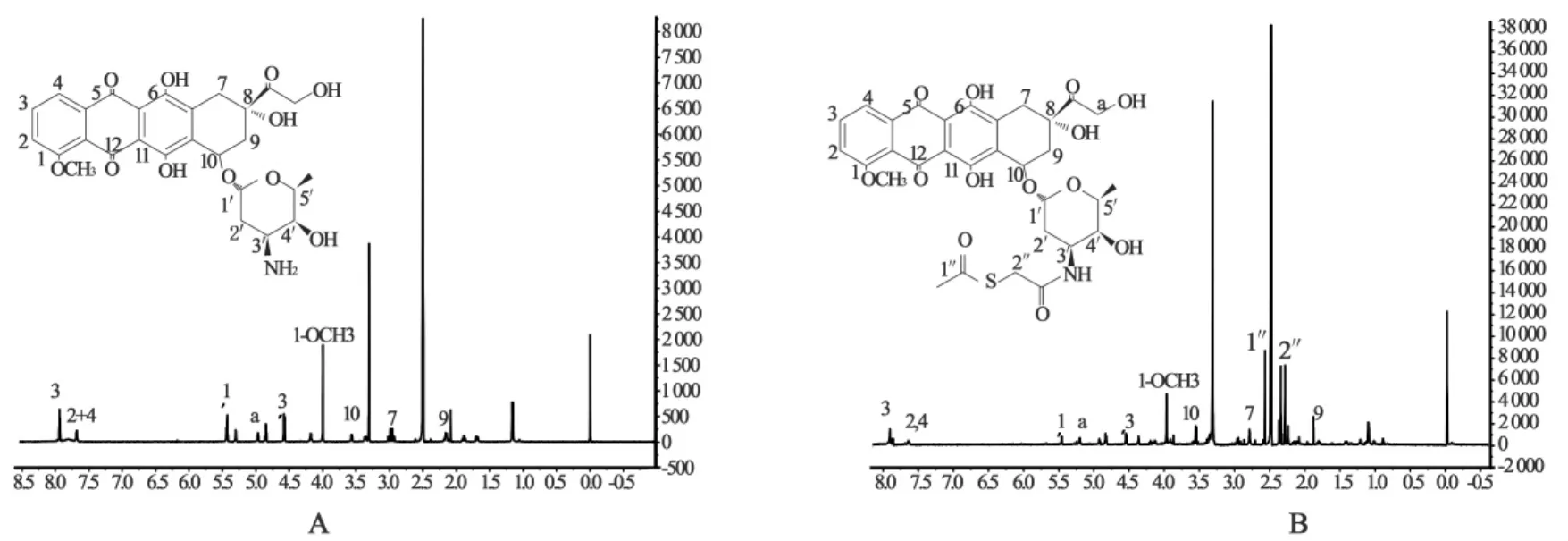
图8 DOX(A)、DOX-SATA(B)的600 M1H NMR谱图
4 讨论
阿霉素分子结构中具有活性氨基,较易与其他试剂形成共价键从而被修饰成具有特定官能团的阿霉素衍生物,并且被修饰的阿霉素抗肿瘤活性没有明显改变。本研究探索了对阿霉素进行巯基化修饰的两种化学合成方法,通过HPLC和MS-ESI等手段对合成的巯基阿霉素进行表征和鉴定。
1H NMR确证DOX-SATA出现了与硫酯基团相连的质子信号,表明新合成的化合物中含有硫酯基团。HPLC和MS-ESI结果显示,两种方法均能合成巯基阿霉素。然而,2-IT试剂法中,由于接枝到阿霉素游离氨基上的2-IT试剂中含有活性较大的亚胺基,亚胺基极易与反应初始阶段形成的游离巯基上的氢结合形成氨气,而母环最终形成环化巯基阿霉素。因此,该方法合成的巯基阿霉素不稳定,随着时间延长巯基阿霉素转变为环化巯基阿霉素,不能满足用作模型药物的游离巯基的要求。SATA试剂法合成巯基阿霉素的反应过程中,首先形成稳定的乙酰硫代乙酸酯取代的阿霉素,然后再在盐酸羟胺的作用下脱去乙酰基团,形成含有游离巯基的巯基阿霉素,该过程中不存在其他易发生的副反应。我们的研究结果表明,SATA试剂法合成含有游离巯基的巯基阿霉素的方法更为可行。
[1]Li PY,Lai PS,Hung WC,et al.Poly(l-lactide)-vitamin E TPGS nanoparticles enhanced the cytotoxicity of doxorubicin in drug-resistant MCF-7 breast cancer cells[J].Biomacromolecules 2010,11(10):2576-2582.
[2]Kibria G,Hatakeyama H,Harashima H.Cancermultidrug resistance:mechanisms involved and strategies for circumvention using a drug delivery system[J].Arch Pharm Res,2014,37(1): 4-15.
[3]Thierry AR,Vige D,Coughlin SS,etal.Modulation of doxorubicin resistance in multidrug-resistant cells by liposomes[J]. FASEB J,1993,7(6):572-579.
[4]Kumar SA,Peter YA,Nadeau JL.Facile biosynthesis,separation and conjugation of gold nanoparticles to doxorubicin[J]. Nanotechnology,2008,10(49):495101.
[5]Barraud L,Merle P,Soma E,etal.Increase of doxorubicin sensitivity by doxorubicin-loading into nanoparticles for hepatocellular carcinoma cells in vitro and in vivo[J].JHepatol 2005,42 (5):736-743.
[6]Ma P,Mumper RJ.Anthracycline nano-delivery systems to overcomemultiple drug resistance:a comprehensive review[J].Nano Today,2013,8(3):313-331.
[7]Wang X,Cai X,Hu J,et al.Glutathione-triggered“off-on”release of anticancer drugs from dendrimer-encapsulated gold nanoparticles[J].JAm Chem Soc 2013,135(26):9805-9810.
[8]Gu YJ,Cheng J,Man CW,et al.Gold-doxorubicin nanoconjugates for overcoming multidrug resistance[J].Nanomedicine,2012,8(2):204-211.
A com parison study of synthesizing methods of thiolated doxorubicin
WU Shan1,ZHANG Yeye1,GUO Haixia2,LIU Junjie1,SUN Zhiguo1,ZHONG Yanqiang1,ZOU Hao1(1.Departmentof Pharmaceutics,School of Pharmacy,Second Military Medical University,Shanghai200433,China;2.NO.266 Hospital of PLA,Chengde 067000,China)
Objective Toinvestigate the optimalmethod for synthesizing thiolated doxorubicin.MethodsThiolated doxorubicin was synthesized through two differentmethods.Doxorubicin was reacted with 2-iminothiolane(2-IT)and S-acetylthioglycolic acid N-hydroxysuccinimide ester(SATA),respectively.The synthesized thiolated doxorubicin was further characterized by HPLC and MSESI techniques.Several factors includingmolar ratios aswell as reaction timewere evaluated.ResultsThe results showed that thiolated doxorubicin could be synthesized via both of the twomethods successfully.Thiolated doxorubicin could be stable when doxorubicin was reacted with SATA.But the crude thiolated doxorubicin could be cyclized easily when doxorubicin was reacted with 2-IT.ConclusionThiolated doxorubicin prepared with SATA ismore feasible than thatwith 2-IT.
thiolated doxorubicin;2-iminothiolane(2-IT);S-acetylthioglycolic acid N-hydroxysuccinimide ester(SATA)
O621.3 [文献标志码]A [文章编号]1006-0111(2014)06-0428-06
10.3969/j.issn.1006-0111.2014.06.008
2014-08-02
2014-10-13[本文编辑]李睿旻
国家自然科学基金项目(30801441);科技部重大新药创制专项(2014ZX09J14107-01B).
吴珊,硕士.Tel:13817291343,E-mail:wushan_1012@ 163.com.
邹豪,副教授.Tel:(021)81871287;E-mail:haozou@ smmu.edu.cn.
猜你喜欢
陕西师范大学学报(自然科学版)(2022年5期)2022-11-09
广州化学(2022年4期)2022-09-01
生物技术通报(2021年12期)2021-02-10
云南化工(2020年4期)2020-05-19
科海故事博览·中旬刊(2020年3期)2020-03-15
商品与质量(2019年31期)2019-11-28
塑料助剂(2019年1期)2019-04-17
安徽化工(2018年5期)2018-10-23
医学研究杂志(2015年5期)2015-06-10
中国药理学通报(2014年2期)2014-05-09
