
电喷雾离子源中样品离子化能量转移理论的初探
2014-05-08 11:14张维冰高方园关亚风张玉奎
色谱 2014年4期
张维冰, 高方园, 关亚风, 张玉奎
(1.华东理工大学,上海200237;2.中国科学院大连化学物理研究所,辽宁大连116023)
电喷雾离子源(ESI)是目前液相色谱-质谱联用最常用的接口,作为一种软电离方式,可直接测定热不稳定的极性化合物、形成多电荷离子,在蛋白质组研究中具有独特的优势。由于可产生带多电荷的分子离子,电喷雾离子源适于研究蛋白质等生物大分子。在生物[1-3]、医疗[4,5]、食品[6-8]、药物[9-11]、环境监测[12,13]等研究领域已得到广泛的应用。
液滴中的电荷来自于不同分子的带电形态,在电喷雾电离中往往形成多电荷离子。关于从带电液滴形成气相离子的过程,目前已有多种理论从不同的角度说明样品的离子化行为。其中有最早由Iribarne和 Thomson[14]提出的离子蒸发机理(ion evaporation model,IEM),由 Dole 等[15]和 Iavarone等[16]提出的带电残基机理(charged residue model,CRM)以及近几年由 Konermann 等[17,18]提出的一项新理论——链弹射理论(chain ejection model,CEM)。
一般认为小分子物质由液相向气相转移的过程遵循离子蒸发机理。这类分析物在溶液中有机酸的协助下,经质子化过程带电,当加在带电液滴上的电场足够大时,表面电荷的静电斥力大于溶剂对离子的引力,分析物离子离开液滴表面。
带电残基理论适用于大的球状物质,例如天然折叠蛋白。在中性水溶液中,大部分蛋白呈现紧凑的球状,电荷及极性基团在球体外部以达到最大的亲水效果。非极性部分在球体内侧,形成不与溶剂接触的疏水内芯。Konermann等[17]认为,经过连续库仑爆炸,最终形成只含有单个分析物的液滴,当溶剂层蒸发殆尽,液滴的电荷转移到分析物分子进而形成气相离子。经过该机理产生的带电离子的电荷量与同等体积带电液滴在瑞利极限状态下的电荷量相等。
变性的蛋白构象高度无序,在电喷雾离子化过程中遵循另一种不同的机理,被称为链弹射理论[17,18]。蛋白变性后,被隔离在内部的非极性基团暴露在溶剂中,分子构型由紧凑亲水变为松散疏水。高度展开的蛋白质疏水的特性使得它不再适宜驻留在液滴内部,迁移至液滴表面,链的末端进入气相。紧接着蛋白质剩余部分按照顺序逐步排出直至与液滴分离。
本文通过对各种分子离子化过程进行分析,基于能量最低原理,提出了离子化能量转移理论,将已有的3种理论加以简化分析,为了解离子化的真实过程,从而选择合适的质谱条件,提高不同样品在质谱中的响应提供了一种可能的参考依据。
1 液滴库仑爆炸过程
一般认为在电喷雾离子源中,样品溶液经过电喷雾离子源离子化的过程包括:(a)在毛细管尖端形成带电液滴;(b)通过连续溶剂蒸发和库仑爆炸,形成极小、高度带电的液滴;(c)气相离子的形成。在毛细管处通上若干千伏的电压,在电场作用下,液体在毛细管管口呈锥形(被称为泰勒锥)[19]。锥尖液体受到库仑斥力作用不稳定,很快破碎成带电液滴;液滴进一步破裂,形成更小的液滴,最终形成气相离子。在整个喷雾与分裂过程中,喷雾室内的环境维持稳定,假设周围电导率一致,对应于其中的某一点的热力学性质不变。从热力学角度对液滴库仑爆炸和形成气相离子的过程进行能量分析。
假设泰勒锥产生的液滴半径为R,de la Mora等[20]提出电喷雾产生的液滴半径与液滴密度ρ、流速Vf、溶剂表面张力γ有关,即:

随着液滴溶剂的蒸发,如果没有表面电荷的作用,液滴将进一步变小,直到全部蒸发。实际上,在液滴表面,为达到最低的静电能,电荷均匀分布。对于带电球形液滴,能承受的最大电荷量为瑞利极限时的电荷量(QR)。
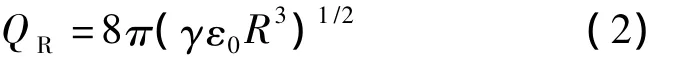
式中ε0为真空介电常数。
由于变形或者电荷极化,液滴在实际稳定状态下所带的电荷量Q可能低于瑞利极限,则球形液滴总能量E可表示为:
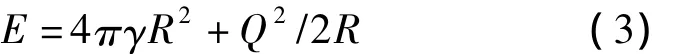
式(3)的右侧第一项为液滴表面能,第二项为静电能[21]。显然,随着液滴半径的变小,静电能不断增大。液滴变小,导致表面能减小,但同时静电能增大。两条曲线的交点为液滴可能存在的最大半径。小于这一半径,液滴蒸发引起的表面收缩将不能够减少整个液滴体系的能量,此时液滴将发生库仑爆炸。
液滴发生库仑爆炸的过程与机理经过了一系列的理论和实验研究。
Ryce和Wayman[22]观测到带电水滴在石蜡油中分裂成不对称的两个液滴。而基于核裂变的液滴理论大部分认为液滴库仑爆炸产生均匀的子液滴。
那么经过库仑爆炸,母液滴究竟是分散成若干个均匀的子液滴,还是两个半径不同的子液滴呢?Ryce和Patriarche[21]从能量的角度对库仑爆炸过程进行了详细的探究。假定:爆炸前后无电荷损失;电荷再分配时间极短,可以忽略;周围环境介电常数均一;那么,液滴在两种情况下可能达到不稳定状态:(1)由于电荷极化使液滴变形,导致液滴所带电荷量低于Q时不稳定;(2)液滴充电迅速,电荷量超出Q。对比产生多个均匀的子液滴和两个不对称的子液滴两种情况下库仑爆炸前后的能量变化,根据能量最低原理,当处于高瑞利带电状态时,倾向于产生多个均匀的子液滴;而处于低瑞利带电状态时,更倾向于产生两个不均匀的子液滴(见图1)。
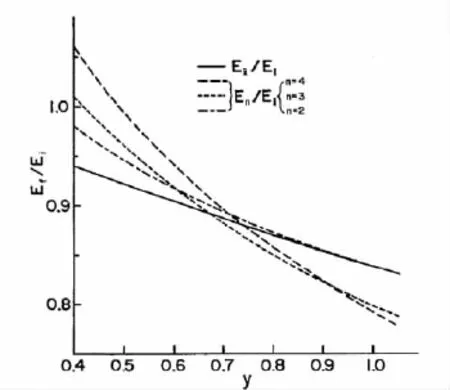
图1 产生2、3、4个均匀液滴和产生两个能量最小、不对称的液滴爆炸前后的能量比Ef/Ei对y(y=Q2/16πγR3)的关系[21]Fig.1 R elationship of the ratio of final to initial energy(Ef/Ei)as a function of y(y=Q2/16πγR3)when dividing sample droplet into two,three,and four symmetric droplets and into two minimum-energy asymmetric droplet[21]
1994 年,Gomez 和 Tang[23]首次利用闪光影像技术成功拍摄到电喷雾离子源中液滴爆炸的过程,同时利用相位多普勒测速技术(PDA)得到爆炸过程中液滴尺寸的分布。最初发生库仑爆炸时液滴所带电荷数Q0≈10-14C,半径为1.5 mm。母液滴产生库仑爆炸前发生变形,形成一个锥突喷射子液滴。实验结果表明,在发生库仑爆炸时,液滴表面电荷量未达到QR,爆炸后子液滴大小相同,直径约为母液滴的1/10。由于瑞利模型是基于以下假设提出的,即表面电荷可移动,因而导致液滴表面电势能均一[24]。因此,由于电荷极化效应或其他原因导致的母液滴严重变形,可能是发生库仑爆炸时液滴表面电荷量未达到QR的原因[25]。
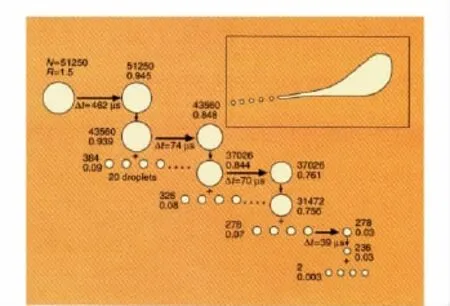
图2 带电液滴库仑爆炸过程示意图[26]Fig.2 Schematic of Coulomb fission of charged droplets[26]The number beside the droplets give radius R(μm)and number of elementary charges N on droplets,Δt corresponds to the time required for fission occurs[23].
此外,Taflin等[25]的研究结果显示实际液滴爆炸过程伴随质量损失(1.0%~2.3%)和电荷损失(10%~18%)。随着库仑爆炸的进行,液滴越来越小,溶剂蒸发速率越来越快,爆炸过程越来越短,导致多普勒测速仪难以拍摄到更小液滴的形成过程。另外,由于缺乏同步光源,无法观测到瞬时现象,因此更小液滴的爆炸过程无法确定是对称爆炸(由一个液滴爆炸为两个相等大小的子液滴),还是像图2所示的那样始终发生非对称爆炸。但无论是对称爆炸,还是由于电场作用造成液滴的扭曲产生不对称爆炸,其过程皆是多种力竞争的结果。当液滴较大时,扭曲可能非常明显,通过扭曲来降低整个液滴的能量;但是当液滴足够小时,表面的曲率半径已经很小,甚至为微尺度,扭曲过程中在增加局域电荷密度的同时,表面积也会相对增加,因此更大的可能为产生对称爆炸。使用更高分辨的拍摄技术,借鉴Vertes等[27]使用染料溶液的荧光发射光作光源,也许可以得到纳米级微液滴的爆炸行为,证实所提出的理论。
根据现有结论,在各种分析物形成气相离子的过程中,均伴随小分子离子或溶质离子等蒸发的过程。除此之外,大分子气相离子的形成过程中还存在电荷转移的现象。把不同种类分析物及不同流动相组成情况下的雾滴爆炸及离子化行为看作不同能量竞争的结果,分析不同电荷转移过程的能量变化和转移。通过研究不同离子的蒸发能、相同或不同离子间的电荷交换能量的变化,以及高能气体分子的碰撞及能量转移对液滴的爆炸及离子蒸发产生的影响,探讨不同电荷转移过程的能量变化和转移特征。为简化模型,现做出如下基本假设:
(1)在整个喷雾与分裂过程中,喷雾室内的环境维持稳定,对应于其中的某一点的热力学性质不变。
(2)带电粒子或者溶质的浓度很小,在液滴不是很小的情况下,不会影响到溶剂的蒸发过程。
2 小分子蒸发的能量转移过程
母液滴经过一系列的库仑爆炸逐渐生成极小的液滴。液滴中的带电小分子离子主要受到(1)溶剂极性相当于电荷把离子拉回液滴和(2)液滴表面电荷形成的电场对带电离子的库仑斥力将离子推离液滴两种作用力。在短距离内引力大,随着距离的增加,引力比斥力递减的快。Iribarne等[14]提出离子蒸发的过程可以用过渡态理论来表示:
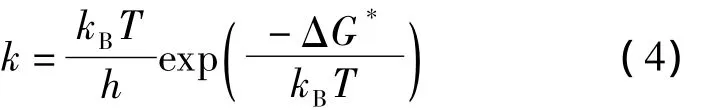
式中:-ΔG*为自由活化能垒的高度,kB为玻尔兹曼常数,h为普朗克常数,T为温度。
现以Na+、K+、NH+4等带单电荷的离子为例,从能量的角度对小分子离子蒸发的过程进行分析。
2.1 小分子离子蒸发的开始
假设库仑爆炸阶段液滴产生n个子液滴(n=R3/r3,其中R为母液滴的半径,r为子液滴的半径,n=2,3,……)。库仑爆炸前系统能量E见式(3),爆炸后系统最终能量 E'见式(5)[21]。
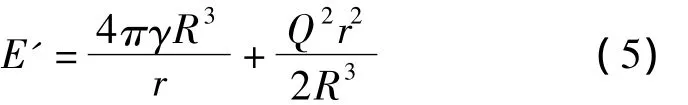
由于离子蒸发产生的气相离子极小,不具有宏观状态下的表面势能及静电能,离子蒸发基本不影响液滴体积,因此发生离子蒸发后系统的最终能量EIEM为:
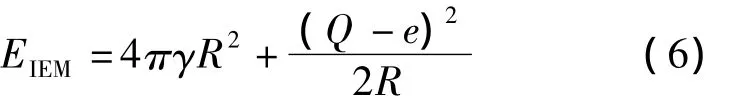
式中e表示元电荷。
一般认为当液滴尺寸小于20 nm[14]时,小分子离子开始蒸发成为气相离子。从能量最低的角度考虑,如图3a所示过程,当液滴较大时,应存在E'<EIEM,此时带电液滴发生库仑爆炸生成子液滴;随着液滴半径的减小,两种情况的能量差值减小;直至R≤20 nm时,EIEM≤E',开始发生离子蒸发。
2.2 小分子离子蒸发的过程
离子蒸发的过程是离子蒸发能与静电能竞争的结果。离子蒸发能G包括两部分[28]:与液滴表面电势有关的ΔG(φ)和溶剂化自由能-Δ,即:
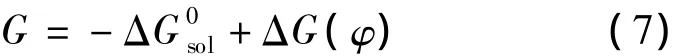
式中 ΔG(φ)是关于液滴表面电势 φ 的函数[14],ΔG(φ)≌Ne2/R。根据库仑定律:
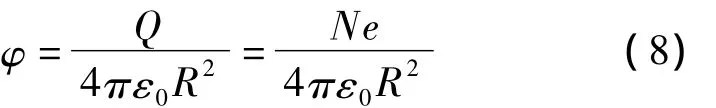
其中:R为液滴半径,N为液滴表面元电荷个数。
假设液滴半径为R,对于即将离开液滴表面的小分子离子,比如 Li+、Na+、K+等,首先是表面电荷中的一员。若不发生离子蒸发,随着溶剂的蒸发,液滴表面的电荷密度增加,离子受到的静电能增大。此时液滴表面电荷量不变,半径变小为r,蒸发前后静电能差为:
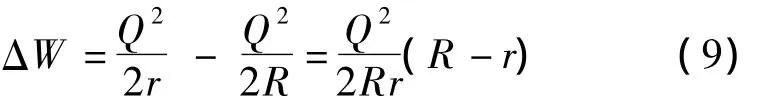
如图3b过程所示,随着溶剂蒸发,液滴逐渐变小,静电能差逐渐增大。当离子蒸发能大于静电能差,即G>ΔW,根据能量最低原理,此时发生溶剂蒸发;随着蒸发的进行,液滴半径减小,ΔW 逐渐增大,当液滴半径减小到某一值时,将出现ΔW >G,此时离子将从液滴表面蒸发。这是离子蒸发理论描述的过程。
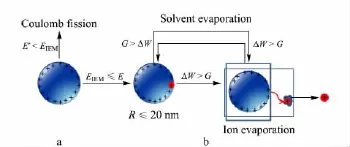
图3 离子蒸发机理示意图Fig.3 Schematic of energy transfer during ion evaporation processa.When R≤20 nm,the ion begins to evaporate.b.When ΔW>G,the ion prefers to evaporate.
3 多电荷离子的形成
Konermann 等[17,18]从分子动力学模型和实验角度均验证了CRM和CEM两种机理(见图4)。选用肌红蛋白为样品,实验结果表明:当蛋白未变性,结构紧凑,离子化过程遵循CRM 理论;而当蛋白质在酸性条件下变性后,结构展开,离子化过程遵循CEM理论。
现从能量的角度,以未变性的蛋白大分子为例,对球状大分子的离子化过程进行分析。对于未变性的蛋白质,电荷暴露在外层形成亲水外壳,非极性基团埋在内部,形成疏水内核[17]。由于分子外层呈现亲水性,溶剂完全蒸干前分析物分子都包覆于液滴内部。带电液滴首先发生库仑爆炸,直至液滴内只含有一个生物大分子。
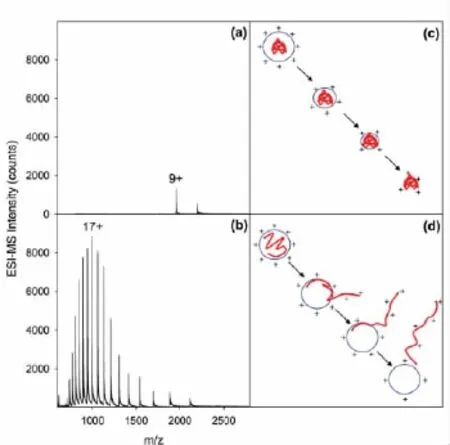
图4 不同大分子在ESI中离子化机理示意图[17]Fig.4 Illustration of ionization mechanisms in ESI for different macromolecules[17]a and b.ESI mass spectra of myoglobin recorded at pH 7 and pH 2,respectively.c and d.the formation of a gas phase macromolecule via CRM and CEM,respectively.
3.1 大分子离子形成过程中的离子蒸发
考虑到蛋白质的半径普遍小于20 nm[29],因此在大分子离子化过程中很有可能伴随表面电解质离子的离子蒸发过程。如图5a过程所示,即当溶剂蒸发至蒸发前后静电能差大于离子蒸发能(ΔW>G)时,小分子离子离开液滴进入气相。此过程重复进行,直至分子表面仅存极薄的溶剂包裹,此时表面离子与分析物分子距离极小。
3.2 多电荷离子的形成
实验[16]证明,在形成气相分析物离子的最终阶段,液滴表面的电荷可转移至分析物分子上,形成多电荷离子。因此认为,在此阶段表面离子不仅存在静电能差ΔW和离子蒸发自由能G,还存在电荷转移能Qtrans,3种能量共同影响离子的行为。所提出的电荷转移能Qtrans指电荷由小分子离子转移至液滴内大分子上所需的能量,在电荷转移发生前,由于高斯定理,位于液滴内部的分子呈现电中性。
通过比较3种能量,根据能量最低原理,即可得知电荷的去向。如果G <ΔW,G<Qtrans,则表面离子蒸发进入气相;若ΔW <G,ΔW <Qtrans,则液滴溶剂继续蒸发;如果Qtrans<G,并且Qtrans<ΔW,根据能量最低原理,表面离子的电荷将转移至分析物分子,形成多电荷离子。这就是带电残基理论描述的过程(见图5b)。
de la Mora[29]证明通过带电残基理论形成的多电荷离子,所带电荷量与相同体积下带电液滴瑞利极限时的电荷量相等。因此可认为,当表面电荷与分析物分子之间距离相对较大时,Qtrans与ΔW 和G有数量级之差,因此不会发生电荷转移;而在仅剩极薄溶剂包裹分析物分子时,表面电荷与分析物表面之间的距离很小,此时普遍存在Qtrans<ΔW 和Qtrans<G,表面离子不再蒸发,电荷完全转移至分析物分子。分析物与表面电荷之间的距离可以极大地影响电荷转移能。
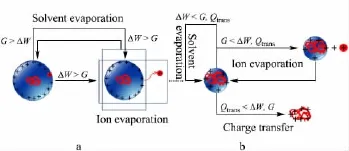
图5 球状生物大分子离子化过程中多电荷离子的形成机理Fig.5 Formation of multi-charged globular macromolecule during ionization processa.When ΔW >G,ion on the surface evaporates.b.When ΔW <G and ΔW <Qtrans,solvent evaporates;when Qtrans<G and Qtrans<ΔW,charges transfer to analyte forming multiple charged ion.
4 链状大分子的链弹射行为
对于大分子链状结构分析物,其支链可分为带电和不带电两种。带电荷的部分可看作离子,不带电的部分类似折叠状态的生物大分子。对链状大分子进行动力学模拟时,Konermann等[17]提出了如下分子模型“+000+0000+000x0+00000+0000+”(+表示带正电荷的侧链,0表示不带电荷,x表示若干个不带电的支链),现在采用该模型来描述我们的能量转移理论。由于化合物带有非极性基团,具有疏水特性,因此分析物首先向液滴表面移动。
4.1 链端的翘起
由于链端带电,这部分的热力学研究可按照离子蒸发理论处理。在离子化过程中,首先链端的离子进入表面,成为表面电荷的一部分;随着溶剂的蒸发,静电能增大,当静电能差ΔW >离子蒸发能G时,离子将从液滴表面蒸发,端基自液滴表面翘起(如图6a所示)。
4.2 长链连续排出液滴的过程
随后支链接近表面电荷,若该支链不带电,则其受到3种能量的共同作用:电荷转移能Qtrans、离子蒸发能G和静电能ΔW。当Qtrans<ΔW 和Qtrans<G时,表面电荷将转移至支链上,使支链带电,溶剂继续蒸发。当该段支链的离子蒸发能G<ΔW和G<Qtrans时,支链从液滴表面蒸发,离开液滴。如果该段支链的Qtrans>ΔW 和Qtrans>G时,无法发生电荷转移,该支链位于液滴表面,占据了液滴表面一定的面积;又由于该链不带电,因此该支链所占据的表面无电荷,液滴表面原有的电荷均匀分布于未被覆盖的液滴表面;与带等量电荷的液滴相比,其电荷密度稍大。假设液滴表面电荷量为Q,半径为R,液滴表面积为4πR2,该支链占据的表面积为 Schain。则表面被疏水链占据前后液滴表面的电势为E1=静电能为由此明显看出 W>2W1。为了追求系统的能量最低,表面电荷倾向于将不带电的支链排出液滴表面(如图6b所示)。
而如果支链带电,类似过程如图6a。当带电支链静电能差ΔW>离子蒸发能G时,从液滴表面蒸发;不带电支链或发生电荷转移后静电能大于离子蒸发能,支链从液滴表面蒸发,或受到表面电荷的作用,被排斥离开液滴。长链离子逐步离开液滴,直至整个链状分子离子进入气相(如图6c所示)。
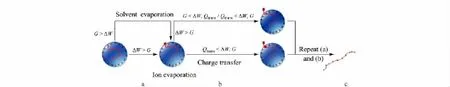
图6 链状大分子离子化过程机理示意图Fig.6 Schematic of the ionization process of a chainlike macromoleculea.The end part:when ΔW > G,side chain evaporates.b.The side chain in the middle part:if the branched chain is charged,the ionization process happened as in Fig.6a.For uncharged side chain:when Qtrans<ΔW and Qtrans<G,the charges transfer to the side chain,then the ionization happens as in Fig.6a;when Qtrans>ΔW and Qtrans>G,the side chain is excluded by charges from the droplet.c.The analyte is gradually excluded out of the droplets after a series of steps a and b in Fig.6.
5 溶剂、电解质离子的综合作用
根据文献描述,除仪器本身的影响外,样品本身的性质可以影响电荷转移能:分子构型能够显著影响表面电荷的分布,松散的结构往往具有更高的带电状态,而结构紧凑的物质带电较少。Ahadi和Konermann[17]比较了 pH=2和 pH=7时肌红蛋白在ESI-MS中的信号强度。X-射线结果表明,pH=7时分子结构紧凑,疏水基团包裹在内部,电荷位于外层;而pH=2时,蛋白质结构松散,整体呈现疏水性。ESI-MS结果显示:pH=7时信号极低,而pH=2时信号强度有了显著增强(增大一个数量级)。另外,分析物的大小同样能影响表面电荷的转移。不只是蛋白质、多肽等生物大分子,实验证明,能产生通过带电残基理论形成多电荷离子的有机物最小的相对分子质量为100。此外,溶剂和电解质同样可以影响电荷转移能。
溶剂的pH、表面张力等性质在决定多肽、蛋白质等生物分子的带电状态中起到了至关重要的作用。Le Blanc等[30]证明pH可影响电喷雾中多肽的带电情况。Chalt等[31]也证实不同pH下蛋白质带电状态不同,不同负离子条件下蛋白质和多肽带电情况也不相同。另外,具有较高表面张力的液滴能够容纳更多的电荷;表面电荷密度越大,转移给分析物的电荷就越多[21]。实验证明,在加入某些特定的改性溶剂,比如甘油、3-硝基苯甲醇后,蛋白质和多肽能够取得较高的带电状态,带电量的增加可能是由于液滴表面张力的增加而引起的。
Konermann等[32]提出使用不同的电解质会造成质谱信噪比的差异。当除H+外,还存在Na+等金属离子为电荷载体时,CRM产物为[M+(ZR-i)H+iNa]ZR+(i=0,1,…,ZR;ZR为电荷数),M为蛋白质分子,H表示因质子化所带电荷。由于Na+与蛋白质结合度不均匀,从而降低了分析物的信噪比。而采用作为电荷载体时,由于最后阶段可变为NH3挥发,因此不会影响信噪比;而且甚至可以替换结合的金属离子,从而提高信号响应。
6 结论
电喷雾离子源质谱是生物分子常用的分析方法,特别适合于分析生物大分子。有关离子化过程,目前得到普遍认可的有3种机理。电荷转移能的提出将3种机理结合,不仅能够解释多电荷的形成、溶剂效应等问题,对于明确电喷雾离子化机理、提高分析物离子化效率、增强响应值均有重大意义。
[1] Eberlin L S,Ferreira C R,Dill A L,et al.Biochim Biophys Acta,2011,1811(11):946
[2] Yew J Y,Cody R B,Kravitz E A.Proc Natl Acad Sci USA,2008,105(20):7135
[3] Chen L C,Nishidate K,Saito Y,et al.Rapid Commun Mass Spectrom,2008,22(15):2366
[4] Cooks R G,Manicke N E,Dill A L,et al.Faraday Discuss,2011,149:247
[5] Girod M,Shi Y,Cheng J X,et al.Anal Chem,2011,83(1):207
[6] Zhang L Y,Wang Z C,Zhang W B.Chinese Journal of Chromatography(张凌怡,王智聪,张维冰.色谱),2013,31(2):122
[7] Zhang Z,Ren F,Zhang P.Chinese Journal of Chromatography(张忠,任飞,张盼.色谱),2012,30(11):1108
[8] Cao P,Mou Y,Gao F,et al.Chinese Journal of Chromatography(曹鹏,牟妍,高飞,等.色谱),2013,31(9):862
[9] Lanekoff I,Thomas M,Carson J P,et al.Anal Chem,2013,85(2):882
[10] Zhou Z,Zhang J,Zhang W,et al.Analyst,2011,136:2613
[11] Chen Y,Zhu J,Yu Z S,et al.Chinese Journal of Chromatography(陈跃,朱军,于忠山,等.色谱),2012,30(11):1148
[12] Gu H,Xu N,Chen H.Anal Bioanal Chem,2012,403(8):2145
[13] Zhang X,Wang N,Zhou Y,et al.Anal Methods,2013,5(2):311
[14] Iribarne J V,Thomson B A.J Chem Phys,1976,64(6):2287
[15] Dole M,Mack L L,Hines R L,et al.J Chem Phys,1968,49(5):2240
[16] Iavarone A T,Williams E R.J Am Chem Soc,2003,125(8):2319
[17] Ahadi E,Konermann L.J Phys Chem B,2011,116(1):104
[18] Konermann L,Rodriguez A D,Liu J.Anal Chem,2012,84(15):6798
[19] Taylor G.Proc R Sot Lond A,1964,280(1382):383
[20] de la Mora J F,Navascues J,Fernandez F,et al.J Aerosol Sci,1990,21(1):S673
[21] Ryce S A,Patriarche D A.Can J Phys,1965,43(12):2193
[22] Ryce S A,Wyman R R.Can J Phys,1964,42(11):2185
[23] Gomez A,Tang K.Phys Fluids,1994,6(1):404
[24] Chung J K,Consta S.J Phys Chem B,2012,116(19):5777
[25] Taflin D C,Ward T L,Davis E J.Langmuir,1989,5(2):376
[26] Kebarle P,Tang L.Anal Chem,1993,65(22):972
[27] Nemes P,Marginean I,Vertes A.Anal Chem,2007,79(8):3105
[28] Kebarle P.J Mass Spectrom,2000,35(7):804
[29] de la Mora J F.Anal Chim Acta,2000,406(1):93
[30] Le Blanc J C Y,Wang J,Guevremont R,et al.Org Mass Spectrom,1994,29(11):419
[31] Mirza U A,Chait B T.Anal Chem,1994,66(18):2898
[32] Konermann L,Ahadi E,Rodriguez A D,et al.Anal Chem,2013,85(1):2
猜你喜欢
地震研究(2021年1期)2021-04-13
分析化学(2020年8期)2020-08-21
高校化学工程学报(2020年2期)2020-06-10
原子能科学技术(2020年1期)2020-03-30
分析化学(2018年4期)2018-11-02
分析化学(2018年7期)2018-09-17
中国学术期刊文摘(2016年1期)2016-02-13
中国地震(2015年1期)2015-11-08
原子能科学技术(2015年1期)2015-03-20
应用技术学报(2014年3期)2014-02-28
