
植物激素样品前处理方法的研究进展
2014-05-08 11:14吴大朋段春凤关亚风
色谱 2014年4期
吴 倩, 王 璐, 吴大朋, 段春凤, 关亚风,*
(1.中国科学院大连化学物理研究所,中国科学院分离分析化学重点实验室,辽宁大连116023;2.中国科学院大学,北京100049;3.中国科学技术大学化学系,安徽合肥230026)
植物激素是一类植物自身合成的痕量化合物。其主要功能是参与调控植物的整个生长过程,包括种子休眠、萌发、营养、生长、生殖、成熟到衰老以及一些对外界的应激反应等,对植物的每一个生命过程都起着重要作用[1]。通过调控植物激素的代谢可显著改良作物株型,提高作物的产量或品质[2]。准确检测植物激素在植物内的浓度变化以及组织特异性分布,是了解其代谢途径和运输过程的重要前提,而这就要求发展准确高效的定性定量分析方法。
植物激素主要包括生长素、赤霉素(gibberellins,GAs)、细胞分裂素、脱落酸、茉莉酸、水杨酸、油菜素甾醇(brassinosteroids,BRs)以及多肽激素[1]。检测主要有3个难点。一是有些重要激素的含量极低,通常都在ng/g甚至pg/g的数量级。不同的激素含量分布也比较广,如生长素、细胞分裂素、脱落酸、茉莉酸和水杨酸一般都在1~100 ng/g鲜重[3],而赤霉素的含量则较之低一个数量级[4]。含量最低的油菜素甾醇通常含量在0.01~0.1 ng/g鲜重[5]。如此低的含量要求其检测方法和仪器对植物激素具有超高的灵敏度。二是植物激素一般不稳定、易分解、对温度和介质环境比较敏感[6]。如赤霉素类激素对环境pH十分敏感,通常在偏酸性条件下才比较稳定[1];同时赤霉素对于温度也十分敏感,温度超过40℃就会发生快速分解[7]。三是植物样品基质十分复杂,直接检测会受到大量基质干扰物的影响。因此对样品处理方法的选择性和除杂能力有很高的要求[8]。
目前植物激素的检测方法非常多,主要可分为生物鉴定法、免疫分析法和色谱分析法3类[8]。生物鉴定法是一种非常经典的植物激素检测分析法。例如油菜素内酯(brassinolide)的水稻叶弯曲测试法(rice-lamina inclination test)可达到0.05 ng/mL的检测限[5]。但它只是一种半定量方法,而且其耗时长,对检测条件要求严苛。一般的化学分析实验室难以实现。而免疫分析法近年来显示出较低检测限,但是其仍需解决制备的抗体的交叉反应问题[9,10]。目前,色谱法应用最广[11],可同时检测多种植物激素[12],且定量准确[13]。近年来,色谱与质谱联用技术快速发展,对植物激素的选择性检测能力有极大提高,能够实现准确定性[14]。
对于色谱法而言,样品前处理是其最为重要的环节之一。前处理决定着分析的准确性和精密度,也是整个分析过程中耗时最长的步骤。而且,植物激素含量低,易分解且基质复杂,这对样品前处理的选择性富集能力,以及操作条件都提出了更高要求。
植物激素的化学特性是决定其前处理方法的重要因素。按极性差异,植物激素可简单分为3类。第1类是中极性植物激素,包括生长素、赤霉素、脱落酸、细胞分裂素、茉莉酸和水杨酸等,多是弱酸性。其他为碱性,统称为酸性或碱性植物激素。第2类是强极性植物激素,主要就是植物多肽类,它们都是由氨基酸组成的肽段,因此为两性化合物,极性很强。而第3类就是弱极性植物激素,主要为油菜素甾醇类,它们都是中性化合物,没有电离能力。以下就以此3类来分别介绍植物激素前处理方法的研究进展。
1 酸性或碱性植物激素
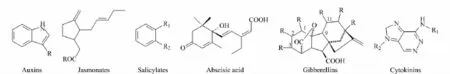
图1 酸性或碱性植物激素的主要结构Fig.1 Structures of acidic and basic phytohormonesAuxins:R=COOH,CH2COOH,CH2CH2CH2COOH,CH2COOCH3.Jasmonates:R=OH,OCH3,NHCH(COOH)CH(CH3)C2H5.Salicylates:R1=COOH,COOCH3,CH=CHCOOH;R2=H or OH.Gibberellins:R=H or OH,positions C1-2,2-3,9-11 are single bond or double bond.Cytokinins:R1=CH2CH=C(CH3)CH2OH,CH2CH2CH(CH3)CH2OH,CH2CH=C(CH3)CH3,CH2-phenyl,CH2-phenyl-OH,CH2-furan;R2=H,β-D-ribofuranosyl,β-D-ribofuranosyl-5'-monophosphate,β-D-glucopyranosyl.
生长素是最先被发现的一类植物激素,为一系列吲哚衍生物,主要包括吲哚-3-乙酸、吲哚-3-丁酸等,主要功能是调节植物生长和细胞分裂等[1];赤霉素是一类四环二萜羧酸类化合物,包括已发现的136种类似物,主要功能是调节植物的生长发育,包括种子萌芽、茎和叶的生长以及开花[1,15];脱落酸是一种单环倍半萜类化合物,主要功能是调节植物种子萌芽和气孔闭合[16];茉莉酸是十二碳的环戊烷酮酸,主要作为植物受到外界伤害时产生的抵御信号[1,17]。它们的基本结构如图1所示。其物理化学性质如表1所示。由此可见,虽然这些激素的功能各不相同,但其化学性质很类似,都为相对分子质量100~400之间的小分子弱酸。因此大部分色谱分析法可以对其做到同时检测。前处理通常会利用它们的弱酸性和极性进行分离。而细胞分裂素则是一类氮杂环的碱性小分子,主要功能是推动细胞分裂、种子发芽以及芽的形成等过程,其极性与前面的弱酸性植物激素类似[1,18]。

表1 主要植物激素的物理化学性质Table 1 Physical and chemical properties of major phytohormones
1.1 样品提取
植物样品大多为固态,需要通过浸提将分析物转移到液态溶液中,方便后续分析操作。浸提的过程比较单一,对于弱酸弱碱类植物激素,通常采用甲醇、异丙醇或乙腈与水的混合溶液进行浸提[19-21]。为了防止分析物的分解或氧化,通常在4℃甚至更低温度下进行[11,19],且浸提液中需加入少量的酸类化合物。如改良的 Bieleski溶剂[22],采用甲醇/甲酸/水(15∶1∶4,v/v/v)的溶液进行浸提,可同时萃取生长素、细胞分裂素、脱落酸和茉莉酸。Pan等[3]采用异丙醇/水/浓盐酸(2∶1∶0.002,v/v/v)的溶液对水稻叶进行浸提,再用液液萃取除杂,通过液相色谱/质谱联用建立了赤霉素(gibberellin 3(GA3)和gibberellin 4(GA4))、吲哚乙酸、脱落酸、水杨酸和茉莉酸等18种内源性有机酸类植物激素的检测方法。另外还有报道通过旋蒸除去有机相,再进行冻融来除去大部分的脂溶性色素或蛋白[7,20]。
1.2 纯化与富集
由于植物基质的复杂性,在进行色谱分析前,通常都需要对浸提液进行纯化或富集。纯化富集的方法通常有固相萃取、固相微萃取、液相萃取和液相微萃取等。
1.2.1 固相萃取
固相萃取(solid phase extraction,SPE)是运用最为广泛的样品纯化除杂方法。根据弱酸类植物激素的极性进行纯化,常用的固相萃取柱为C18柱、亲水亲油平衡柱(Oasis HLB)等[12,23]。C18 柱可以除去大量非极性的叶绿素基质,同时也可以通过调节淋洗液和洗脱液来除去一些极性很强的基质。但其除杂能力有限,当植物中含有大量非极性基质时常常会产生很大的基质干扰峰。所以单纯采用C18柱仅仅能适用于一些基质不太复杂的样品。如Ma等[24]采用C18柱对椰汁进行除杂,联用液相色谱/质谱可同时对生长素、脱落酸、赤霉素进行检测,回收率可达到67%~95%。Oasis HLB是一种亲水亲油平衡柱,尽管其极性较大,但单独使用也难以达到满意的净化效果。通常将它与液液萃取等方法联用来提高除杂效率[25]。根据弱酸类植物激素的酸碱性,近年来有人将离子交换作用引入到纯化过程中。Ge等[26]采用一根反相和阴离子交换混合机制的固相萃取柱(OasisMAX)对椰汁中的11种赤霉素进行了萃取,可达到500倍左右的富集倍数和80%左右的回收率。为进一步提高选择性,一些高选择性的材料也被用于植物激素的萃取,如分子印迹材料[27,28]。Yan 等[28]采用分子印迹技术对香蕉中的生长素进行了测定,萃取相对回收率为78%~107%,体现了萃取方法较好的选择性。
很多时候,单根柱的萃取方法难以处理复杂的植物组织提取液,因此多种小柱串级萃取方法的应用更为广泛[12,29],C18 与 OasisMAX 联用萃取、Oasis HLB 与 C18联用[30]、Oasis HLB 与 OasisMAX 联用[13,23]都有报道。
近年来发展起来的基质固相分散萃取(matrix solid-phase dispersion,MSPD)是一种用于固体和黏稠液体样品的前处理技术[31],它实际上是把前面提及的固相萃取过程和浸提过程同时进行,使得提取和净化步骤合二为一[32]。MSPD首先利用机械混合研磨时产生的剪切力使样品破裂完全,加快溶出、提取,再利用样品基质与化学固定相选择性相互作用得到初步分离。它将样品基质与固定相材料混合研磨,并填装到固相萃取柱中,再用溶剂将分析物洗脱。该法具有简便、灵活、快速、低耗等优点,已被广泛应用于药物[33]、农药[34]、天然成分以及复杂植物[35]和动物[36]样品分析。我组将MSPD 用于拟南芥样品中赤霉素分析的前处理,并结合液相色谱-串联质谱(HPLC-MS/MS)进行检测,建立了MSPDHPLC-MS/MS测定拟南芥中痕量赤霉素GA1、GA3和GA4的分离检测方法,且成功地用于拟南芥地上部分中GA1、GA3和 GA4的测定[37]。其处理流程如图2所示:首先将拟南芥样品与C18填料混合研磨制成MSPD柱,并采用80%冷甲醇洗脱。然后采用反相C18色谱柱进行分离,以0.05%甲酸水溶液和乙腈为流动相进行梯度洗脱,采用电喷雾(electrospray ionization,ESI)负离子模式电离,多反应监测模式检测。对样品前处理条件、色谱分离条件和质谱检测条件进行了优化,结果表明,在最优条件下,3种赤霉素在10~300 ng/g范围内呈良好线性,相关系数(r)均大于0.99,检出限在1.1~4.1ng/g之间。在10~50 ng/g添加水平下,平均回收率范围为54.7%~102.6%,相对标准偏差(relative standard deviation,RSD,n=3)在3.2% ~12.8%之间。该方法操作简单、灵敏度高、选择性好、回收率高,适合拟南芥中GA1、GA3、GA4的含量测定。
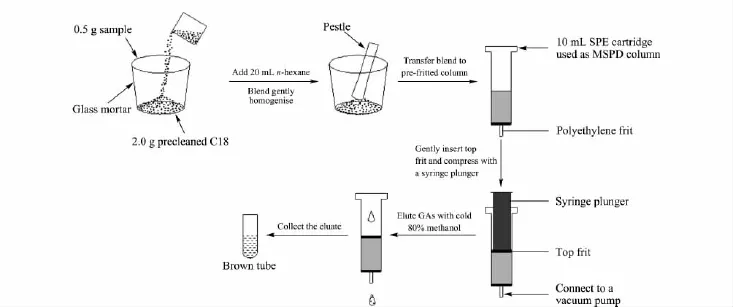
图2 基质固相分散处理植物样品分析赤霉素的示意图Fig.2 Schematic illustration of MSPD method for analysis of gibberellins in plant sample
1.2.2 固相微萃取
固相微萃取(solid phase microextraction,SPME)是一种微型化的固相萃取方法,样品富集和纯化可同时完成,具有较大的富集能力。其溶剂消耗量少,是一种绿色环保的萃取方法[38]。但固相微萃取的样品回收率过低,通常不到60%,对于微量存在于植物中的激素需要大量的植物样品进行富集[39]。而且由于萃取材料容量过低,需要材料有较高选择性,否则由于基质效应,萃取回收率会大幅降低,所以早期应用于基质复杂和含量低的植物激素前处理上的研究较少。但近年随着质谱检测灵敏度的逐步提高,也开始有一些应用报道。如Liu等[40]采用商品化的Carbowax涂层的固相微萃取柱对吲哚乙酸和脱落酸等激素进行了选择性萃取。Liu等[18]采用(2-丙烯酰胺基-2-甲基-1-丙磺酸)-(乙烯基二甲基丙烯酸)共聚物(poly(AMPS-co-EDMA))合成的整体毛细管柱直接分离植物组织中的细胞分裂素,其萃取回收率在标样中可达95%以上,但在实际样品中仅能达到59.1%~77%。可见其基质效应较大。
1.2.3 液相萃取
液相萃取在传统方法中通常与固相萃取联用来提高纯化效果[4,8,12,41]。但随着质谱检测灵敏度和选择性的提高,也有直接对液液萃取后的提取液进行质谱检测的报道。Pan等[3]采用二氯甲烷对浸提液进行液液萃取除杂后就直接进行液相色谱/质谱检测,但检测到的激素只是含量较高的生长素、脱落酸、茉莉酸等。
1.2.4 液相微萃取
以上的讨论表明:对于基质复杂(如植物组织)且含量低的酸性植物激素(如赤霉素),单级固相萃取或微萃取纯化方法难以满足痕量植物激素分析检测的需求。其实,大部分固相萃取材料都以疏水作用为主导,即使引入离子交换或使用分子印迹材料,材料上的有机官能团也会带来较大的疏水非特异性作用,这样对植物中大量的脂质和色素的去除很不利。而传统的多级固相微萃取又具有步骤多、繁琐耗时的缺陷。液液液微萃取方法是一种将液相萃取和反萃取结合的微型液相萃取方法。它以样品相和接受相的pH梯度为传质动力,中间有机相的组成决定萃取选择性,非常适合于萃取有电离能力的有机化合物[42]。近年来中空纤维膜的引入使得这种方法可以达到与固相微萃取相近的稳定性,应用更为简便[43-46]。Wu 等[47]将中空纤维膜液液液微萃取(hollow fiber-based liquid-liquid-liquid micro extaction,HF-LLLME)应用到椰子汁中水杨酸、茉莉酸、吲哚乙酸、脱落酸的萃取,绝对回收率在8%~43.7%,富集倍数为48~243倍。但作为一种微萃取方法,其回收率过低的问题非常突出,所以并不适合样品量有限的植物样品前处理,仅适合这类样品量大的果汁样品。
我组发展了水渗透增强中空纤维膜液液液微萃取方法,并应用于赤霉素的萃取分析[48]。水渗透过程由提供相(即样品相)和接受相之间的离子强度差造成。接受相内水向外渗透同时浓缩了分析物,进一步增加了富集倍数并最终突破平衡常数,其原理如图3a所示[48]。将这种改进的液液液微萃取应用到赤霉素萃取富集中,首先将植物的80%甲醇浸提液经抽滤、旋蒸、冷冻、离心处理后作为液液液微萃取的供给相,采用中空纤维微滤膜孔中充满的有机溶剂作为有机相,膜的内腔注入碱性水溶液作为接受相,将膜放入处理后的植物浸提液中进行萃取[7]。装置如图3b所示。我们采用单因素实验法对中空纤维膜中有机相组成和样品相盐度进行了优化,采用正交试验对提供相和接受相pH值、萃取温度进行了优化,通过传质模拟对萃取时间和接受相体积进行了优化。将优化后的条件作为最优的方法测定了水稻中的8种赤霉素,提高了萃取方法对赤霉素类化合物的选择性。结果表明,对不同赤霉素,萃取富集倍数为100~800倍不等,萃取回收率在10%~80%之间[7]。对8种赤霉素的检测限可达1.6~61 pg/mL,相对标准偏差在10%以下。采用这种方法检测了不同时期的水稻样品中8种赤霉素的含量。
现在很多全自动的固相萃取或微萃取仪都已商品化,但由于液相微萃取的稳定性较差,其通量和自动化研究不多,商品化极少。
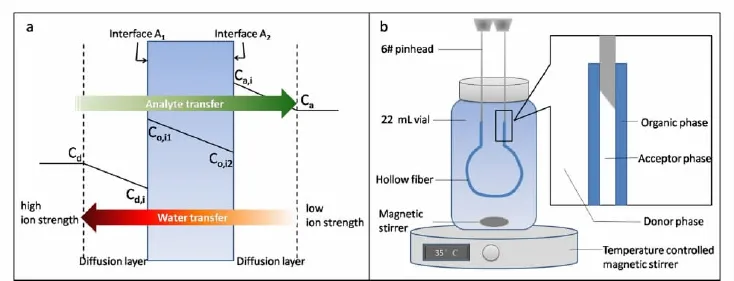
图3 水渗透液液液微萃取(a)传质过程示意图[48]和(b)装置示意图Fig.3 (a)Schematic description of mass transfer process[48] and(b)the device of HF-LLLME with water osmosis
2 油菜素甾醇
油菜素甾醇类化合物是植物体内调控植物生长的一类相当重要的植物激素[6]。其分子结构如图4所示,其物理化学性质见表1。但它的检测难度更大。首先,其含量极低(成熟的茎和叶中仅0.01~0.1 ng/g鲜重[6]),另外,缺乏具有高灵敏检测响应的特征官能团[6,9]。即使在灵敏度和选择性极高的液相色谱/质谱联用分析仪上,由于它没有可电离基团,在电喷雾离子源上的响应仍相当微弱。Gamoh等[49]首次采用大气压化学电离源(atmospheric pressure chemical ionization,APCI)对萘硼酸衍生化的油菜素甾醇进行检测,仅能达到2~4 ng的检测限。Swaczynova等[10]采用ESI离子源对油菜素甾醇进行检测,对几种特殊的油菜素甾醇类化合物能达到50 fmol(约25 pg)的检测限。为了进一步提高检测灵敏度,多采用衍生法来提高油菜素甾醇对于不同检测器的响应。但目前适合于液相色谱分析的前处理方法还很少,且存在很多问题[50-52]。主要是因为常规前处理方法没有足够的选择性,油菜素甾醇回收率低、杂质残余多、基质效应大,难以满足极低浓度分析物的检测要求。
2.1 样品提取
和前面的酸性植物激素不同,油菜素甾醇的极性比较小,通常采用80%的甲醇-水溶液在4℃进行浸提[51,53,54]。也有文献采用纯的甲醇或乙腈进行浸提[52,55,56]。
2.2 纯化与富集
2.2.1 固相萃取
油菜素甾醇处于中极性区,植物色素和脂肪酸的干扰更加严重,要求除杂更加彻底。传统样品前处理方法需要经过两步以上的液相萃取,再用串级固相萃取来达到纯化的目的[51,57-59]。近年来,由于新的固相萃取材料的引入,这个过程得以少许简化。Xin等[54]采用反相阴离子交换混合机制柱(MAX)和反相阳离子交换混合机制柱(MCX)进行两级SPE,除去大量的可电离化合物,得到的中性化合物再用硼酸键合的磁性纳米球除去糖类中性化合物,测定的brassinolide(BL)和castasterone(CS)两种油菜素甾醇的回收率达70%左右,硼酸磁性纳米球除去糖类后,基质效应大大减小,灵敏度提高了5倍。

图4 油菜素甾醇类化合物的结构Fig.4 Structures of brassinosteroids
虽然串级的固相萃取柱可以有较高的回收率,但这种基于除杂的纯化方法往往有较大的基质效应,而且过程繁琐、损失也大。于是有人利用具有更强选择性的固相萃取材料,如分子印迹或亲和材料对油菜素甾醇进行选择性萃取,同时一步排除杂质[52,56]。早在 1994 年,Gamoh 等[52]就采用硼酸亲和凝胶(affin601)对植物提取液中的油菜素甾醇进行选择性萃取,萃取回收率可达90%以上,但其由于相互作用太强使得洗脱非常困难,需要向洗脱液中加入5%的过氧化氢才能洗脱90%,之后很少有人使用这种方法。
2.2.2 固相微萃取
对于油菜素甾醇类的萃取纯化来说,采用固相微萃取方法的研究并不多,这主要是由于其含量太低,样品绝对回收率必须很高才能检出。Pan等[60]采用丙烯酰胺-乙二醇二甲基丙烯酸酯共聚物(poly(acrylamide-co-ethylene glycol dimethacrylate),poly(AM-co-EGDMA))涂层修饰的固相微萃取纤维针对油菜素甾醇进行萃取,证明聚合物涂层对油菜素甾醇有一定的选择性,相对回收率在70%~104%,并检测到花粉中的油菜素甾醇(0.6~1.9 μg/g)。
2.2.3 在线微固相萃取
为了进一步提高油菜素甾醇萃取方法的选择性和萃取效率,利用油菜素甾醇的邻二羟基与硼酸之间的亲和作用以及其较强的疏水作用,本课题组[56]发展了一个具有二维机制的油菜素甾醇在线微固相萃取(two dimensional online micro solid phase extraction,2D-online μSPE)前处理方法。将亲和填料与疏水填料串联,亲和填料通过亲和基团与BRs分子中的邻二羟基基团发生亲和作用,而疏水填料则通过疏水链与BRs分子中的碳环发生疏水作用。这两种填料完全不同的作用机制和作用位点使得它们有很好的正交性,因此两种填料分段填充的SPE微柱对BRs具有很高的选择性。另外,疏水填料在硼酸填料后端,对BRs与亲和填料强亲和作用而产生的宽洗脱谱带,在疏水填料段被压缩在端头区,然后置换冲洗溶液,压缩馏分的谱带,达到高倍数富集。其作用原理如图5所示。同时两种填料的对接可实现第一维填料洗脱的同时就是对第二位填料的上样,省去多级串联SPE小柱操作的繁琐性和损失,实现在线全自动化的处理。

图5 在线二维微固相萃取-在柱衍生前处理系统对油菜素甾醇分析的原理图[56]Fig.5 Schematic drawing of 2D-online μSPE for brassinsteroid analysis[56]
该装置可以实现在30 min内完成植物粗提液的选择性萃取和富集,并在线衍生,最后通过中心切割给液相色谱上样。这套体系与液相色谱-三重四极杆质谱联用,对检测的5种油菜素甾醇(BL,CS,6-deoxo-24-epicastasterone (d-epiCS),teasterone(TE)和 typhasterol(TY))的检测限达到 0.1 pg/mL。另外,此方法定量相当准确,且基质效应极小。因此可以直接用外标法定量,避免采用标准加入法而浪费植物样品,也不需要使用同位素标准品控制基质效应。外标法测定的相对回收率在80%~120%之间,RSD小于10%。与HPLC-MS/MS联用,番茄叶中的5种油菜素甾醇均能检出,检出和加标谱图如图6所示。
2.3 衍生反应
油菜素甾醇结构中既没有紫外响应的芳香基团或共轭基团,也没有ESI源响应的可电离基团,更没有荧光响应基团,因此化学增敏衍生对于油菜素甾醇化合物的检测必不可少。近年来油菜素甾醇衍生方法的研究较多,大多基于硼酸与油菜素甾醇结构中的邻二羟基之间的特异性反应。Svatos等[61]采用丹磺酰氯-3-氨基苯硼酸(dansyl-3-aminophenylboronate)对油菜素甾醇进行衍生,其在ESI源下的标样检测限能达到0.1 pg。Huo等[53]开发了一种新的衍生试剂2-溴吡啶-5-硼酸(2-bromopyridine-5-boronic acid),对油菜素甾醇进行衍生,衍生过程比以前的方法更为简单,不需要加热和长时间的反应,仅需数秒钟振摇即可完成衍生反应,达到2~8 pg/mL的检测限。Xin等[54]采用6-甲氧基-3吡啶硼酸(6-methoxy-3-pyridinylboronic acid,MPyBA)对油菜素甾醇进行衍生,其在标样中的检测限为1 pg左右。
以上衍生反应都在试管内进行,难以在线应用,且在线过程中也会存在谱峰扩散或溶剂不兼容的问题。结合前面提到的二维固相微萃取前处理装置,我们组[56]发展了一种对油菜素甾醇进行柱上衍生的方法。我们采用氨基苯硼酸作为衍生试剂,在二维SPE的C18柱区段上在柱衍生,然后柱上洗脱,进样阀定量环中心切割衍生产物,最后转阀将纯化浓缩衍生后的BRs进样给HPLC。这种在柱衍生的方法,可以实现衍生试剂的在线浓缩,当采用同样浓度衍生试剂时,在柱衍生比在管衍生的灵敏度提高了3~10倍。当与前面的二维SPE在线系统连接时,其检测限可达0.1 pg/mL。

图6 在线二维微固相萃取-在柱衍生前处理系统分析番茄叶中5种油菜素甾醇的色谱图(225 mg番茄叶组织,浸提液10 mL)[56]Fig.6 Chromatograms of tomato leaf samples(225 mg tissues for each 10 mL sample)after online 2D μSPE-on column derivatization-HPLC-MS/MS analysis[56]
3 植物多肽
植物多肽[62,63]是近20年来才逐步被人关注的一类植物激素。研究表明,一些植物多肽同样参与了植物生长发育的调控以及细胞间的信号传导。多肽激素包括系统素(systemin)、植物硫肽素、S位点富含半胱氨酸蛋白/S位点蛋白质11(S-locus cysteine-rich protein/S-locus protein 11,SCR/SP11)、基因CLAVATA3(CLV3)以及快速碱化因子等[63]。其中仅系统素[64,65]的前处理方法研究较多。
系统素是植物受到外界伤害时在细胞质中产生的具有信号传导功能的内源多肽物质,也是目前植物伤害生理这一新领域研究中最关键的因素[17]。其化学结构包括18个氨基酸,为一类极性很强的两性化合物,对其提取和纯化的方法与前面提到的有机酸类和甾醇类有较大差别。
3.1 样品提取
由于多肽的强极性和两性,其提取通常采用纯的蒸馏水就可以进行[11]。对于含水量较多的样品通常直接匀浆过滤就可以进行纯化。
3.2 纯化与富集
植物多肽具有强极性和两性特点,常用二乙基氨基乙基纤维素(diethylaminoethyl cellulose,DEAE)柱纯化,也有使用强阳离子交换柱与C18柱进行多级萃取的报道[66-69]。同时由于蒸馏水提取,提取液中通常含有大量大分子蛋白,常需用葡聚糖凝胶G25(Sephadex G25)进行过滤除杂。
近年来,Liu等[64]采用免疫亲和柱首次对系统素进行了选择性分离,采用液相色谱-四极杆串联飞行时间质谱(quadrupole coupled of time of flight mass spectrometry,QTOFMS)进行检测。免疫亲和柱是将抗番茄系统素的多克隆抗体共价键合到CNBr活化的琼脂糖凝胶上,将键合好的吸附剂填充到SPE小柱中进行萃取,萃取结果表明该材料对番茄系统素有较高的选择性,实际样品加标回收率能达到90%左右,而对比马铃薯系统素I和II则选择性很差,加标回收率仅30%~50%。
表2总结了3类植物激素近年来主要发展的样品前处理方法。
4 总结与展望
随着色谱/质谱联用技术的发展,前处理方法会变得更加简便。简便、绿色和快速是样品前处理方法的主要追求目标,新颖的前处理方法以及新型的萃取材料和衍生试剂正在不断涌现。

esnrm o o h h ytoree p f thsis o汇an aly总e方法th r理s fo处d的eth 前o素m激植aratio n物类3rep p 2p le表sam e thf oarym m u S2leabT nceefere ]R [24[25];%.4 validation 101-%.5 d 92 etho%M 95-recovery%67 acid recovery gibberellic alytesn A ,abscisic acid gibberellins,auxin ,abscisic acid acidgibberellic atrix m it ple ilk ifru Sam m ut kiw conco hineseC detho cartridge BL H mpreparation 18 C asis O E ple SP Sam Single oneshorm basic ones or lant horm P cidic A phyto[26][28][30][23][37][40][18][48][3][54][52][60][56][61][53][56]][64 500%A).6IB %99%77-ent factor%.6%(-%%.7%%%.311110294.1 94107-;enrichm- -),59104-120-%very %%%.8%%90.0.7(IAA ple%%atrix effects decrease%%34 reco 8078>754%sam 807080 98 recovery L -about recovery recovery recovery.85real - - ,m /m L/m %acid 98in %10%85%70%90pg pg 8pg .9 92 abscisic recovery relative relative relative relative recovery very recovery recovery.10-2.1 0 reco very reco very reco very reco very reco relative relative D O D D L O L O L recovery A and gibberellins L-B AB gibberellins d-CS homo cytokinins,auxins,auxins,abscisic acids ates,auxins,E,,T ,24epid-CS E,,T gibberellins L TY o-BL omS,gibberellins gibberellins cytokinins gibberellins S,S TY auxins on jasm,C SS ins L ,C epi-B,C ,C ,h ,C asterol.B L B 24L B L B L B L B system typhisTY ilk m thaliana es es plants ut leaf leaf cocon ana baoshanensis tissu tissu ban fruit rice arabidopsis viola plant rice plant rice pollen ato tom ato--tom solanaceae teasterone,and sphere E is agnetic m nate-epicastasterone,T X A )18ronicacid acid n A odified m m MA nic -bo asisM B 18 DM ro GD A silica-C-P phenylboro bo ino colu yl 6-deoxo-24 ating-E X X -C -O S is L L B B coS-co A ax-E C -am pyridine-5 asisM H H w MP-M odified o m phen noaffinity-C asis IP ino 18u m arbo asis poly(A X A M O O O C C M ffin601M-co poly(A A A B P dansyl-3-brom2 am im s ESP E n castasterone,d ultiple SP D E E S is M M L E E E M E M SP M SP L L L L SP SP nline μ O derivatizatio SP lide,C rassinosteroids lypeptides brassino isL B B o P
但这些新方法、新材料的真正应用价值仍有待检验。一是某些新方法虽然实现了针对某些样品和某些分析物的简便快速绿色的前处理,但主要是基质不太复杂的样本(果汁等),而且其目标化合物往往是植物中含量比较高的组分(某些酸性植物激素)。其实对于真正具有检测难度的分析物以及样品,则往往仍达不到分析要求。二是某些新方法虽然较传统方法有了更简便的操作过程,或更高的选择性,但其整个处理流程和形式仍没有太大突破;对于植物组织样品,液氮研磨、低温储藏以及长时间的浸提过程仍是必须的。但植物组织破碎、浸提等过程不仅仅耗时长,而且使得检测结果难以真正体现植物激素在植物活体内的空间分布以及时间动态变化,更不能区分激素在植物基质中的存在状态(如游离态或结合态),使得分析方法与植物学检测的前沿需求仍然相距甚远。三是植物学研究通常需要同时测定很多样本,因此分析方法的通量和方法检测精密度仍有待提高。而一些新的萃取材料,如固相微萃取材料通常着重于考察选择性回收率等参数,而缺乏萃取材料制作重复性、方法重现性研究,以及与传统商品化材料的系统比较。
针对植物激素分析需求,笔者认为在以下几个方面应继续展开深入研究:一是对于基质复杂、含量低的植物激素分析,如植物组织叶、茎等部位的油菜素甾醇,应进一步提高样品前处理方法的效率和选择性,以达到少量、微量样品的分析要求。二是针对植物学上对检测分析的前沿需求,发展一些针对活体样品、具有时空分辨率的前处理方法,拓展样品前处理领域。如近年来发展的原位固相微萃取方法,具有很好的活体样品前处理的潜力。三是针对激素在植物组织中的存在状态,发展一些新的材料或方法,实现植物激素的形态分析,如蛋白结合的植物激素和游离态的植物激素。
总之,对于超低含量基质复杂的植物激素的准确、快速、简便的分析,以及活体原位植物激素的动态分析问题,仍然是现在植物激素分析的挑战。而这些问题的解决都离不开前处理技术的深入发展。植物激素定量分析水平的进步,也将推动植物激素相关的植物学基础研究的发展。
[1] Davies P J.The Plant Hormones:Their Nature,Occurrence,and Functions//Davies P J.Plant Hormones.Springer Netherlands,2010:1
[2] Sakamoto T,Morinaka Y,Ohnishi T,et al.Nat Biotechnol,2006,24(1):105
[3] Pan X,Welti R,Wang X.Nat Protoc,2010,5(6):986
[4] Fernández H,Doumas P,Bonnet-Masimber M.Plant Growth Regul,1997,22(1):29
[5] Takatsuto S.J Chromatogr A,1994,658:3
[6] Bajguz A,Hayat S.Brassinosteroids-Occurence and Chemical Structures in Plants//Hayat S,Ahmad A.Brassinosteroids:A Class of Plant Hormone.Springer Netherlands,2011:1
[7] Wu Q,Wu D,Duan C,et al.J Chromatogr A,2012,1265:17
[8] Fu J H,Sun X H,Wang J D,et al.Chinese Sci Bull,2011,56(4/5):355
[9] Khripach V A,Zhabinskii V N,Litvinovskaya R P,et al.Immunoassays of Brassinosteroids//Hayat S,Ahmad A.Brassinosteroids:A Class of Plant Hormone.Springer Netherlands,2011:375
[10] Swaczynova J,Novak O,Hauserova E,et al.J Plant Growth Regul,2007,26(1):1
[11] Du F,Ruan G,Liu H.Anal Bioanal Chem,2012,403(1):55
[12] Hou S,Zhu J,Ding M,et al.Talanta,2008,76(4):798
[13] Izumi Y,Okazawa A,Bamba T,et al.Anal Chim Acta,2009,648(2):215
[14] Fletcher A T,Mader J C.J Plant Growth Regul,2007,26(4):351
[15] Li J,Xiao L T,Zeng G M,et al.J Agric Food Chem,2005,53(5):1348
[16] Vilaro F,Canela-Xandri A,Canela R.Anal Bioanal Chem,2006,386(2):306
[17] Bai Y,Du F Y,Liu H W.Chinese Bulletin of Life Science(白玉,杜甫佑,刘虎威.生命科学),2010,22(1):36
[18] Liu Z,Wei F,Feng Y Q.Anal Method,2010,2(11):1676
[19] Stephan M,Bangerth F,Schneider G.Plant Growth Regul,1999,28(1):55
[20] Nefed’eva E E,Mazey N G.Appl Biochem Microbiol,2009,45(4):454
[21] Chen H,Zhang Z X,Zhang G M,et al.J Agric Food Chem,2010,58(8):4560
[22] Nishiyama R,Watanabe Y,Fujita Y,et al.Plant Cell,2011,23(6):2169
[23] Kojima M,Kamada-Nobusada T,Komatsu H,et al.Plant&Cell Physiol,2009,50(7):1201
[24] Ma Z,Ge L,Lee A S,et al.Anal Chim Acta,2008,610(2):274
[25] Wang J,Zhang H,Hu M,et al.Food Industry(王骏,张卉,胡梅,等.食品工业),2008(4):74
[26] Ge L,Peh C Y,Yong J W,et al.J Chromatogr A,2007,1159:242
[27] Kugimiy A,Takeuchi T.Anal Chim Acta,1999,395(3):251
[28] Yan H,Wang F,Han D,et al.Analyst,2012,137(12):2884
[29] Menendez V,Revilla M A,Bernard P,et al.Plant Cell Rep,2006,25(10):1104
[30] Zhan S X,Sun S H,Zang L N,et al.Chinese Journal of Health Laboratory Technology(湛社霞,孙世宏,臧李纳,等.中国卫生检验杂志),2009,19(3):579
[31] Barker S A.J Chromatogr A,2000,885:115
[32] Capriotti A L,Cavaliere C,Giansanti P,et al.J Chromatogr A,2010,1217:2521
[33] Míguez-Framil M,Moreda-Pineiro A,Bermejo-Barrera P,et al.J Chromatogr A,2010,1217:6342
[34] Sobhanzadeh E,Abu Bakar N K,Abas M R B,et al.J Hazard Mater,2011,186(2/3):1308
[35] Visnevschi-Necrasov T,Cunha S C,Nunes E,et al.J Chromatogr A,2009,1216:3720
[36] Cai X,Wang C,Xu J,et al.J Chromatogr B,2011,879(9/10):657
[37] Wang L,Wu Q,Duan C F,et al.Chinese Journal of Chromatography(王璐,吴倩,段春凤,等.色谱),2011,29(9):923
[38] Pawliszyn J.Solid Phase Microextraction:Theory and Practice.New York:Wiley-VCH,1997
[39] Hyotylainen T,Riekkola M L.Anal Chim Acta,2008,614(1):27
[40] Liu H T,Li Y F,Luan T G,et al.Chromatographia,2007,66(7/8):515
[41] Nakayama M,Yamane H,Yamaguchi I,et al.J Plant Growth Regul,1989,8(3):237
[42] Pedersen-Bjergaard S,Rasmussen K E.J Chromatogr A,2008,1184:132
[43] Kou D,Wang X,Mitra S.J Chromatogr A,2004,1055:63
[44] Yazdi A,Eshaghi Z.Talanta,2005,66(3):664
[45] Huang S.J Chromatogr A,2006,1135:6
[46] Melwanki M,Huang S.Anal Chim Acta,2006,555(1):139
[47] Wu Y L,Hu B.J Chromatogr A,2009,1216:7657
[48] Wu Q,Wu D,Geng X,et al.J Chromatogr A,2012,1248:32
[49] Gamoh K,Abe H,Shimada K,et al.Rapid Commun Mass Spectrom,1996,10(8):903
[50] Gamoh K,Kitsuwa T,Takatsuto S,et al.Anal Sci,1988,4(5):533
[51] Gamoh K,Omote K,Okamoto N,et al.J Chromatogr,1989,469:424
[52] Gamoh K,Yamaguchi I,Takatsuto S.Anal Sci,1994,10(6):913
[53] Huo F,Wang X,Han Y,et al.Talanta,2012,99:420
[54] Xin P,Yan J,Fan J,et al.Analyst,2013,138(5):1342
[55] Fujioka S,Takatsuto S,Yoshida S.Plant Physiology,2002,130(2):930
[56] Wu Q,Wu D,Shen Z,et al.J Chromatogr A,2013,1297:56
[57] Friebe A,Volz A,Schmidt J,et al.Phytochemistry,1999,52(8):1607
[58] Schrick K,Cordova C,Li G,et al.Phytochemistry,2011,72(6):465
[59] Schmidt J,Altmann T,Adam G.Phytochemistry,1997,45(7):1325
[60] Pan J,Hu Y,Liang T,et al.J Chromatogr A,2012,1262:49
[61] Svatos A,Antonchick A,Schneider B.Rapid Commun Mass Spectrom,2004,18(7):816
[62] Lindsey K,Casson S,Chilley P.Trends Plant Sci,2002,7(2):78
[63] Ryan C A,Pearce G.Plant Physiol,2001,125(1):65
[64] Du F Y,Bai Y,Liu H W.Anal Chem,2010,82(22):9374
[65] Xie Y H,Ding Z X,Ou Y,et al.Southwest Horticulture(谢永红,丁志祥,欧毅,等.西南园艺),2006,34(1):45
[66] Pearce G,Strydom D,Johnson S,et al.Science,1991,253(5022):895
[67] Pearce G,Moura D S,Stratmann J,et al.Nature,2001,411(6839):817
[68] Mucha P,Rekowski P,Kupryszewski G,et al.J Chromatogr A,1996,734:410
[69] Pearce G,Bhattacharya R,Chen Y C,et al.Plant Physiol,2009,150(3):1422
猜你喜欢
食品安全导刊(2022年32期)2022-12-07
吉林蔬菜(2022年4期)2022-11-04
今日农业(2021年21期)2021-11-26
今日农业(2021年14期)2021-10-14
许昌学院学报(2018年8期)2018-09-05
娃娃乐园·综合智能(2018年3期)2018-03-22
数学小灵通(1-2年级)(2017年10期)2017-11-08
浙江大学学报(农业与生命科学版)(2015年4期)2015-12-15
新疆农垦科技(2014年7期)2014-02-28
中国粮油学报(2014年8期)2014-02-06
