
集成化蛋白质组定量分析平台的构建及应用
2014-05-08 11:14袁辉明张丽华张玉奎
色谱 2014年4期
周 愿, 张 珅, 袁辉明, 张丽华*, 张玉奎
(1.中国科学院大连化学物理研究所,中国科学院分离分析化学重点实验室,国家色谱研究分析中心,辽宁大连116023;2.中国科学院大学,北京100039)
蛋白质是生命活动的体现者和执行者。因此,在研究生命过程中,通常不仅需要了解哪些蛋白质参与,而且还应该揭示蛋白质的含量及其变化[1]。因此亟须发展高准确度、高精密度和高通量的定量蛋白质组学技术。近年来基于液相色谱-串级质谱联用(LC-MS/MS)的方法得到了快速发展,已成为蛋白质组定量的主流技术。该方法可以分为无标记和稳定同位素标记定量法。其中,基于稳定同位素标记的蛋白质组定量方法可以在不同样品预处理步骤中实现样品混合,进而实现多重标记并消除LCMS/MS分析过程中引入的误差,已成为定量蛋白质组学研究中最常用的方法[2]。
二甲基化标记是一种常用的稳定同位素标记方法[3]。该方法利用甲醛和氰基硼氢化钠及各自的同位素形式标记蛋白质/肽段中所有的活性氨基。具有反应条件温和、快速、高效、无明显副反应、同位素试剂种类多、价格低廉等优点。常用的二甲基化标记多是基于溶液标记,易引起样品的损失而导致定量准确度差。此外,还难以实现样品的自动化分析。为此,Heck等[4]使用具有反相保留机理的预柱对捕集的肽段进行在线二甲基化标记,并和LCMS/MS联用,构建了集成化的蛋白质组定量分析平台。Wang等[5]在此基础上,在RPLC捕集柱上进行在线固相二甲基化标记后,用二维强阳离子交换色谱-反相色谱-电喷雾串级质谱(2D-SCXRPLC-ESI-MS/MS)平台实现了标记肽段的在线分离分析。利用上述平台对蛋白质组样品进行自动化定量分析,不仅降低了手动离线操作带来的误差,而且减少了样品的标记时间。然而这些工作均是肽段水平标记,有可能在肽段水平混合前引入实验误差,不利于蛋白质组的准确定量。由于基于稳定同位素标记的方法中标记样品混合的步骤越是靠前,定量结果越准确[6-8],因而在蛋白质组样品标记后直接混合应优于肽段水平标记后混合的定量结果。
为发展基于蛋白质水平标记后混合的定量方法,蛋白质组的高效在线酶解至关重要。Tian等[9]将蛋白质样品捕集在SCX柱上,然后连续通入胰蛋白酶溶液,实现了蛋白质的在线酶解。然而该方法无法实现复杂蛋白质组样品的分级,且容易导致样品丢失。近年来,我们[10-13]发展了多种由蛋白质在线分离、酶解和肽段分离鉴定构成的集成化蛋白质组分析平台,显著提高了蛋白质组的定性分析能力。
在此基础上,将蛋白质组样品二甲基化后,采用由微柱弱阴/弱阳离子交换色谱(μWAX/WCX)-固定化酶反应器(IMER)-nanoRPLC-ESI-MS/MS构成的平台,实现了蛋白质组的相对定量分析。与常规的肽段水平标记后混合的方法相比,该方法具有高准确度、高通量和自动化的优点。此外,利用该平台实现了小鼠腹水型肝癌淋巴道高、低转移细胞系(Hca-F/Hca-P)的差异蛋白质分析。
1 实验部分
1.1 仪器与试剂
尿素、二硫苏糖醇(dithiothreitol,DTT,纯度99%)、碘乙酰胺(iodoacetamide,IAA,纯度 98%)、胰蛋白酶(trypsin)、甲醛(formaldehyde,CH2O,38%(v/v)水溶液)、氘代甲醛(formaldehyde-d2,CD2O,20%(v/v),溶于 D2O)、氰基硼氢化钠(sodium cyanoborohydride,NaCNBH3)、N-(3-二甲基氨基丙基)-N'-乙基碳二亚胺(N-(3-dimethylaminopropyl)-N'-ethylcarbodiimide,EDC)、N-羟基丁二酰亚胺(N-hydroxysuccinimide,NHS)和乙烯基吡咯烷酮(N-vinyl-2-pyrrolidinone,NVP)(Sigma公司);过硫酸铵(ammonium persulfate,APS,Acros公司);乙腈(acetonitrile,ACN,色谱纯,Merck公司);其他试剂均为国产分析纯。实验用水为Milli-Q纯水系统(Millipore公司)制备的超纯水。WAX和WCX 填料(10 μm,100 nm(1 000 Å),TOSOH 公司),C18填料(5 μm,10 nm(100 Å),Phenomenex公司)。Hca-F/Hca-P由大连医科大学邵淑娟教授惠赠。
实验使用的仪器包括高效液相色谱-电喷雾-线性离子阱-静电场轨道离子阱质谱仪(HPLC-ESILTQ-Orbitrap Velos,Thermo Fisher Scientific公司)、高速离心机(Beckman Coulter公司)、组织破碎仪(Biospec Products公司)、超声破碎仪(Vernon Hills公司)和真空浓缩仪(Thermo Fisher Scientific公司)。
1.2 高、低转移细胞系蛋白质的提取
首先,两种细胞系用1×PBS缓冲液(phosphate buffered saline,pH 7.2)洗净血液,并用含有 0.1%(v/v)蛋白酶抑制剂的8 mol/L尿素溶液作为裂解液悬浮细胞系。将细胞在10 000 r/min的转速下于冰浴中匀浆1 min,然后超声破碎100 s(破碎能量设置为100%)。超声破碎后的悬浮液在4℃冰箱中消泡后,在25 000 g下离心30 min。弃去沉淀,上清液按照样品与丙酮体积比为1∶4的比例加入-80℃丙酮过夜。于1 000 g下离心10 s,弃去上清后冷冻干燥。最后用8 mol/L尿素溶液将蛋白质复溶,并用Bradford法测定蛋白质浓度。样品在-20℃保存待用。
1.3 蛋白质二甲基化标记
向200 μg Hca-P 提取蛋白质中加入 4 μL 0.1 mol/L DTT,在56℃水浴中反应1 h;再加入8 μL 0.1 mol/L IAA,避光反应30 min。稀释10倍后,将其等分成两份分别用16 μL轻标记试剂(4%(v/v)CH2O,0.6 mol/L NaCNBH3)和重标记试剂(4%(v/v)CD2O,0.6 mol/L NaCNBH3)标记1 h;标记完成后,加入16 μL 1%(v/v)氨水终止反应。用捕集柱(C8,10 mm ×4.6 mm,5 μm,30 nm(300 Å))除去标记蛋白质中的盐;冷冻蒸干后于-20℃保存待用。
Hca-F提取蛋白质经类似步骤进行DTT还原和IAA烷基化后,用重标记试剂(CD2O+NaCNBH3)标记,然后和轻标记的Hca-P按照等质量混合。除盐后冷冻蒸干,于-20℃保存待用。
1.4 hIMER的制备
按照文献[14]所述方法制备hIMER。简言之,将10 mg聚丙烯酸酯修饰的氨基微球加入到NVP溶液中(NVP∶H2O=1∶9,v/v),并用 APS 引发,在40℃下反应40 h。用水和甲醇清洗3次后,将微球注入5 cm ×150 μm 的毛细管中。将2.5 mg/mL胰蛋白酶、5 mg/mL EDC和5 mg/mL NHS溶于100 mmol/L磷酸盐缓冲液(pH 5.0),振荡2 h。然后在室温下将混合液不间断地通入毛细管中进行胰蛋白酶的固定,反应时间为6 h。用溶于20 mmol/L NH4HCO3缓冲液(pH 8.0)的25%(v/v)乙腈冲洗后,再用20 mmol/L NH4HCO3缓冲液(pH 8.0)冲洗,然后于4℃保存待用。
1.5 在线酶解和LC-MS/MS分析
将5 μg轻(L)、重(H)标记的 Hca-P/Hca-P 或Hca-P/Hca-F蛋白质混合物分别以1 μL/min的流速上样到 WAX/WCX柱(60 mm ×150 μm)。采用5个盐台阶梯度(20,50,100,200和 1 000 mmol/L的醋酸铵溶液,pH 8.0)将蛋白质洗脱至hIMER中,并进行不停留在线酶解。每个组分的酶解产物用C18的捕集柱捕集后,依次进行nanoRPLCESI-MS/MS分析。
nanoRPLC分离条件如下。C18色谱柱:200 mm×75 μm;流动相 A:98%H2O+2%ACN+0.1%甲酸(FA),流动相 B:98%ACN+2%H2O+0.1%FA;梯度:0~15~15.1~20~100~105~105.1~125 min,0%B~0%B~0%B~5%B~35%B~80%B ~80%B ~0%B;前15 min流速为6 μL/min,后110 min流速为300 nL/min。
质谱条件如下。LTQ-Orbitrap Velos质谱;喷雾电压:2.0 kV;离子传输毛细管加热温度:250℃。全扫描模式分辨率(Rs):60 000,扫描范围:m/z 350~1 800;每个全扫描后选择20个强度最强的离子进行碎裂;子离子扫描采用碰撞诱导解离(CID)模式和数据依赖采集(DDA)模式;碰撞能量:35%;母离子分离窗口宽度:2 Th;采用动态排除模式,重复次数为1,排除时间为40 s。
1.6 数据处理
所有采集到的数据采用 MaxQuant(1.3.0.4版本)内置的Andromeda软件作为搜索引擎。蛋白质序列库选择 IPI.mouse.fasta(3.68 版本,共 56 729种蛋白质序列),并附加反库和常见的污染物蛋白质库。半胱氨酸烷基化(+52.021 5)设为固定修饰;蛋白质的N端乙酰化(+42.061 6)、甲硫氨酸氧化(+15.994 9)设为可变修饰。氨基酸标记设置:轻标记时Lys的相对分子质量增加28.031 3,重标记时Lys增加32.056 4。蛋白酶选择胰蛋白酶,允许最多4个漏切位点。一级质谱质量容差为6 ppm,二级质谱质量容差为20 ppm,肽段和蛋白质水平的假阳性率(FDR)均低于1%。选择蛋白质簇中的唯一性肽段用于蛋白质的定量,至少有1条唯一性肽段。
2 结果与讨论
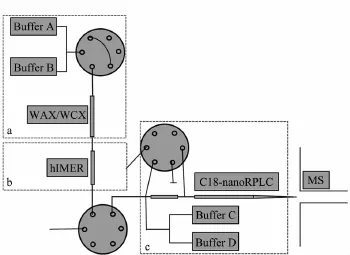
图1 集成化蛋白质组定量分析平台示意图Fig.1 Scheme of integrated platform for proteome quantificationa.μWAX/WCX for online separation of the dimethylated proteins;b.hIMER for online digestion;c.nano-RPLC for separation of the protein digests.WAX/WCX:weak anion exchange/weak cation exchange chromatography;hIMER:hydrophilic immobilized enzymatic reactor.
如图1所示,构建了新型蛋白质定量分析平台。该平台包括以下3个部分:(a)微柱弱阴/弱阳离子交换色谱(μWAX/WCX)用于标记蛋白质的分级;(b)hIMER用于蛋白质水平的在线酶解;(c)nanoRPLC用于酶解产物的分离。通过该集成化系统可实现标记蛋白质的分级、在线酶解和酶解产物的分离。
2.1 在线酶解对蛋白质组定量的影响
蛋白质的酶解效率对蛋白质组定量极为关键。常见的酶解方法可分自由溶液酶解和IMER酶解。与自由溶液酶解相比,IMER的酶解效率更高,但是容易产生含漏切位点的肽段,导致定量结果中离散点较多,从而影响定量准确度[15]。本实验采用经亲水性基团改性的IMER进行在线酶解。将Hca-P蛋白质变性、还原和烷基化后直接进行轻、重二甲基化标记,以H/L=1∶1(质量比)混合后经WAX/WCX分级,并在hIMER上进行不停留酶解后进行分析,考察该平台在线酶解对定量结果的影响。
表1列出了所有肽段及匹配的漏切肽段的定量比值,可以看出共有21组肽段有不漏切和漏切两种情况。将使用漏切肽段获得的H/L结果与使用无漏切肽段获得的结果相比,结果介于0.533和1.37之间,说明采用漏切与无漏切肽段进行的定量结果相近。结果表明,在蛋白质水平标记后混合进入IMER实现同时酶解,可以消除漏切对定量准确度的影响。
此外,考察了hIMER基质的非特异性吸附对定量的影响。尽管采用hIMER可以显著降低基质的非特异性吸附,但仍然不可完全避免。以轻、重标记的KACGDSTLTQITAGLDPVGR为例,考察了该肽段在不同盐浓度洗脱时获得的定量比值。从图2可以看出,在不同盐浓度下该肽段的强度从8.97×106逐渐降低到 1.92×105,而 H/L比值分别为1.18、1.22 和1.16;这说明 hIMER 的非特异性吸附对肽段的定量影响较小。
实验中采用了hIMER在线无停留的酶解,酶解时间仅为10 min。相比于24 h的自由溶液酶解,hIMER酶解大大降低了样品预处理的时间,从而提高了定量分析的通量。
2.2 蛋白质水平的二甲基化标记效率
二甲基化标记通常是在肽段水平上进行的,但由于甲醛与氨基反应效率较高,因此本文利用甲醛与蛋白质中赖氨酸(K)上的氨基反应进行蛋白质水平标记,并考察了标记效率。
以H/L=1∶1(质量比)的Hca-P提取蛋白质为样品,经蛋白质水平的二甲基标记和集成化平台分析,共鉴定到625条肽段,其中440条含有K。含有不同数目K的肽段的标记效率如表2所示。当肽段中含有5个K时,有4条肽段上的K只标记了3~4个,被完全标记的肽段的比例仅为42.9%;在其余情况下被完全标记的肽段的比例均在90%左右。这说明尽管在蛋白质水平的二甲基化标记效率略低于肽段水平,但蛋白质组定量结果与理论值相符。

表1 经hIMER在线酶解产生的肽段及其匹配的漏切肽段的定量结果Table 1 Quantification results of peptides and the corresponding peptides with miscleavages resulting from onlinedigestion by hIMER
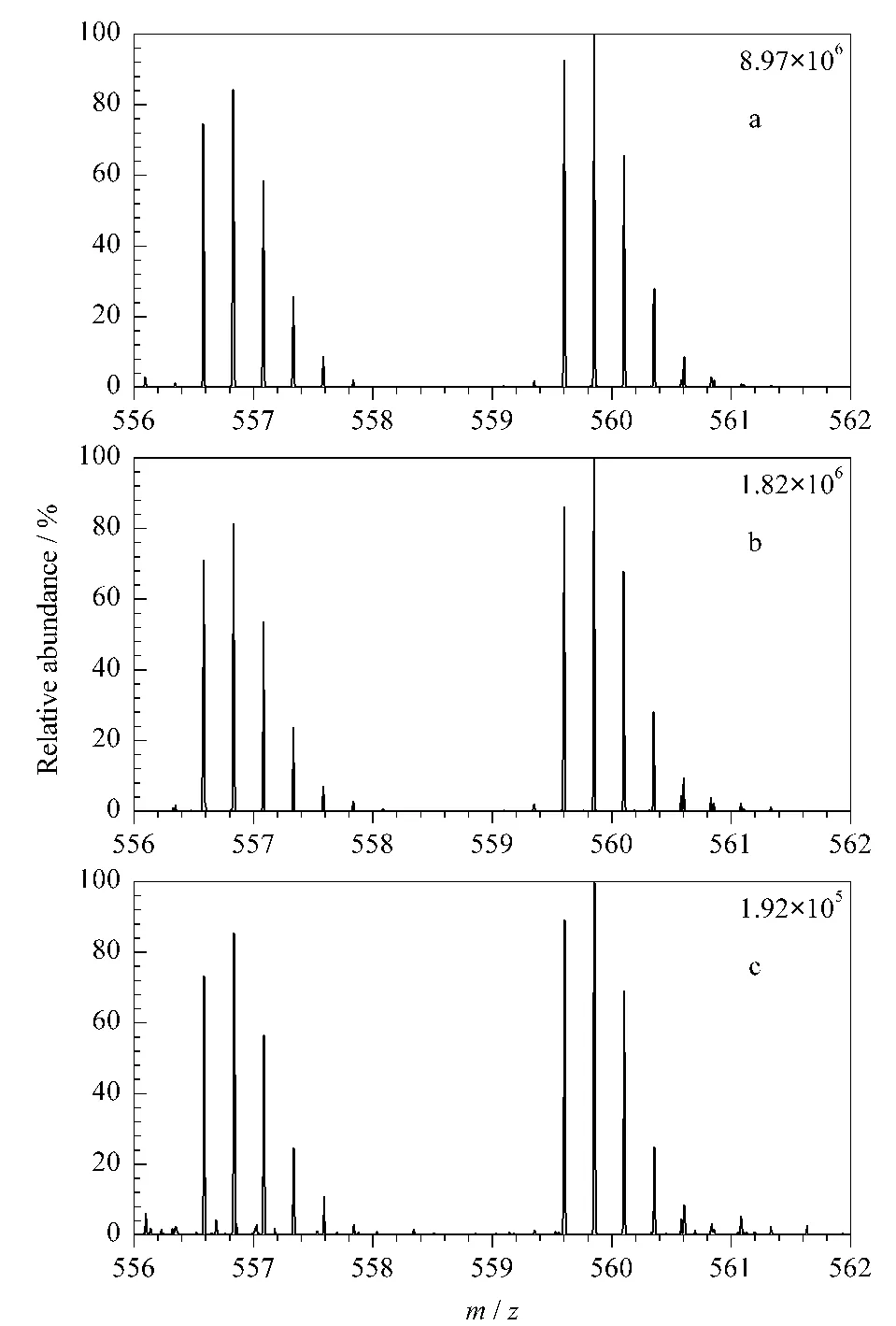
图2 轻、重标记的肽段KACGDSTLTQITAGLDPVGR在不同NH4Ac浓度洗脱后的定量结果Fig.2 Quantification results of H/L labeled peptide(KACGDSTLTQITAGLDPVGR)eluted by different NH4Ac concentrationsa.20 mmol/L;b.50 mmol/L;c.100 mmol/L.

表2 二甲基化蛋白质酶解产物中含有不同赖氨酸数目的肽段的标记数量Table 2 Labeled peptides of dimethylated proteins digests with different lysine numbers
2.3 集成化蛋白质组定量平台的性能评价
以H/L=1∶1(质量比)混合的二甲基化标记的Hca-P蛋白质为样品,平行分析两次;选择两次都定量到的蛋白质进行后续分析,共103种。蛋白质组的定量结果分布如图3所示,获得的H/L的比值介于0.589 和 2.30,平均值为 1.01;说明该平台具有良好的定量准确度。这应归因于蛋白质水平标记混合后进行酶解,消除了漏切和肽段水平混合产生的误差。此外,利用该平台可以实现自动化蛋白质组定量分析,避免了人为操作引入的误差。
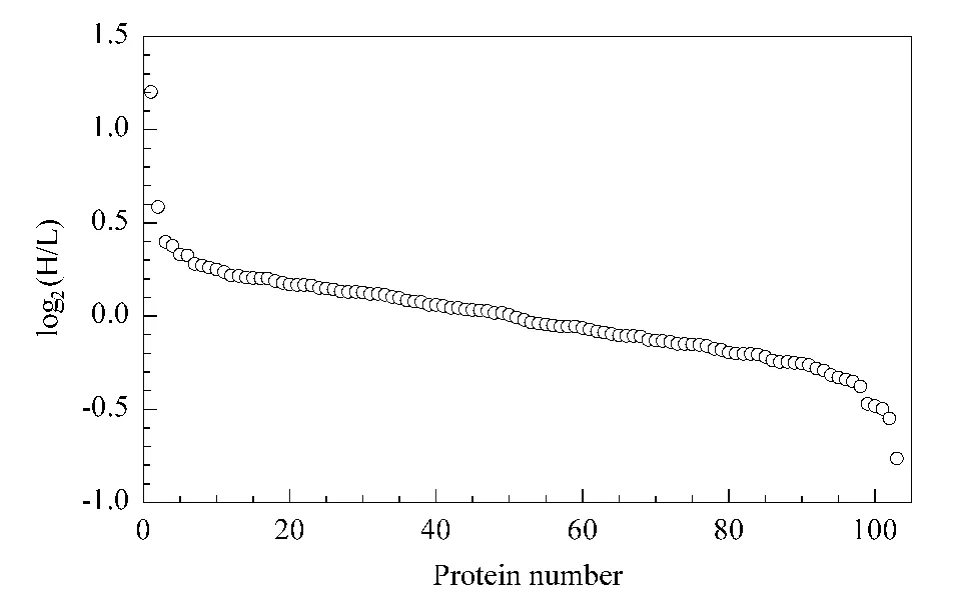
图3 H/L=1∶1(质量比)的Hca-P蛋白质混合样品经集成化定量分析平台得到的蛋白质的比值Fig.3 Protein expression ratios(H/L=1∶1(mass ratio),Hca-P protein mixture)obtained by the integrated quantification platform
2.4 集成化蛋白质组定量平台的应用
利用该集成化蛋白质组定量分析平台,开展了小鼠腹水型肝癌淋巴道高、低转移细胞系的差异蛋白质分析。实验平行分析了3次,选择2次以上定量到的蛋白质进行后续分析,共197种蛋白质;以2倍差异为依据时,共发现12种蛋白质在高转移细胞系中低表达,15种蛋白质在高转移细胞系中高表达。
在高转移细胞系中高表达的蛋白质中,HSP-27是热休克蛋白质的一种,参与抑制NF-αB通路从而阻断细胞凋亡,已在多个实验中被证实在高转移能力的细胞系中显著高表达[16-18];膜联蛋白 A3(Annexin A3)是膜联蛋白的一种,也已被证实该蛋白质的上调与肿瘤的发生、发展与转移等过程密切相关[19]。
在高转移细胞系中低表达的蛋白质中,Fga(fibrinogen,alpha polypeptide isoform 1)、CNBP(cellular nucleic acid-binding protein)和SLIRP(SRA stem-loop-interacting RNA-binding protein,mitochondrial)通过准等重二甲基化标记的方法[17]定量到,且变化趋势与本实验的结果相同。Fga除参与凝血外,还参与细胞-细胞间的黏附以及细胞与基质间的黏附作用;CNBP是一种单链DNA结合蛋白,调节脂类代谢;SLIRP是一种核受体辅阻遏子,与SRA RNA结合,抑制SRA-介导的核受体的共激活。这3种蛋白质抑制癌细胞转移的机理尚未见报道。
3 结论
本文构建了一种由二甲基化蛋白质分级、在线酶解、肽段分离和质谱分析构成的集成化蛋白质组定量分析平台,并将其用于小鼠腹水型肝癌淋巴道高、低转移细胞系的差异蛋白质分析。结果显示,利用该平台显著提高了蛋白质组定量的准确度、通量和自动化程度。因此有望与SILAC技术结合,在规模化蛋白质组定量中发挥重要作用。
[1] Yates J R Ⅲ,Washburn M P.Anal Chem,2013,85(19):8881
[2] Merrill A E,Coon J J.Curr Opin Chem Biol,2013,17(5):779
[3] Boersema P J,Raijmakers R,Lemeer S,et al.Nat Protoc,2009,4(4):484
[4] Raijmakers R,Berkers C R,de Jong A,et al.Mol Cell Proteomics,2008,7(9):1755
[5] Wang F J,Chen R,Zhu J,et al.Anal Chem,2010,82(7):3007
[6] Ong S E,Mann M.Nat Chem Biol,2005,1(5):252
[7] Bantscheff M,Schirle M,Sweetman G,et al.Anal Bioanal Chem,2007,389(4):1017
[8] Zhou Y,Shan Y C,Zhang L H,et al.Chinese Journal of Chromatography(周愿,单亦初,张丽华,等.色谱),2013,31(6):496
[9] Tian R J,Wang S,Elisma F,et al.Mol Cell Proteomics,2011,10(2),DOI:10.1074/mcp.M110.000679
[10] Yuan H M,Zhou Y,Zhang L H,et al.J Chromatogr A,2009,1216(44):7478
[11] Yuan H M,Zhang L H,Hou C Y,et al.Anal Chem,2009,81(21):8708
[12] Yuan H M,Zhou Y,Xia S M,et al.Anal Chem,2012,84(11):5124
[13] Xia S M,Tao D Y,Yuan H M,et al.J Sep Sci,2012,35(14):1764
[14] Jiang H,Yuan H M,Liang Y,et al.J Chromatogr A,2012,1246:111
[15] Wang F J,Wei X L,Zhou H,et al.Proteomics,2012,12(21):3129
[16] Sun M Z,Liu S Q,Tang J W,et al.Proteomics,2009,9(12):3285
[17] Zhou Y,Shan Y C,Wu Q,et al.Anal Chem,2013,85(22):10658
[18] Liu W,Ma Y,Huang L,et al.Mol Biol Rep,2010,37(7):3207
[19] Wu N,Liu S Q,Guo C M,et al.Clin Transl Oncol,2013,15(2):106
猜你喜欢
上海计量测试(2020年1期)2020-03-18
汽车观察(2018年9期)2018-10-23
电子制作(2017年22期)2017-02-02
浙江大学学报(工学版)(2016年2期)2016-06-05
山东医药(2015年14期)2016-01-12
环境科技(2015年1期)2015-11-08
医学研究杂志(2015年4期)2015-06-10
药学与临床研究(2015年4期)2015-06-05
中国医药导报(2015年26期)2015-02-28
中国记者(2014年3期)2014-05-14
