
超高效液相色谱串联质谱法测定人血浆中阿托伐他汀浓度
2014-05-02 03:32吴晓莉杨昌云甘惠贞
中国药业 2014年14期
戴 立,吴晓莉,杨昌云,甘惠贞
(中国人民解放军第180医院药学科,福建 泉州 362000)
阿托伐他汀(atorvastatin,ATV)是具有选择性和竞争性的羟 甲戊二酰辅酶 A(HMG-CoA)还原酶抑制剂,能降低总胆固醇、低密度脂蛋白和三酰甘油的血浆浓度,临床广泛用于治疗冠心病和血脂异常等疾病[1]。阿托伐他汀口服吸收迅速,且存在明显的首关效应,绝对生物利用度仅12%,大剂量服用后可能发生横纹肌溶解等不良反应[2],临床使用过程中发现其在疗效及不良反应方面均存在个体差异,而此现象是否与阿托伐他汀的血药浓度之间存在联系,有待研究证实。为研究阿托伐他汀在人体的药动学过程及治疗药物监测的需要,本试验中建立了灵敏度较高的超高效液相色谱串联质谱(UPLC-MS/MS)法,用于测定阿托伐他汀血药浓度。
1 仪器与试药
Acquity UPLC H-class色谱系统、Xevo TQD质谱仪(Waters公司);Heraeus Pico21离心机(美国赛默飞世尔科技公司);AE240型双量程电分析天平(梅特勒-托利多仪器有限公司);G560E型涡旋混合仪;pHS-3C型精密pH计(上海雷磁仪器厂);HSC-12A型氮吹仪(天津恒奥科技发展有限公司);CQX25-06型超声波清洗器(上海必能信超声有限公司);微孔滤膜(0.22μm,美国津腾公司)。阿托伐他汀钙对照品(批号为23922106,LKT公司,含量99.2%);黄体酮(批号为100027-200307,中国药品生物制品检定所);乙腈、甲酸(Darmstadt Germany Merck KgaA);甲醇、乙酸乙酯、冰乙酸、磷酸二氢钠、氢氧化钠均为分析纯(上海强顺化学试剂有限公司);去离子水(自制)。
2 方法与结果
2.1 标准溶液配制
称取阿托伐他汀钙对照品0.8 mg,精密称定,用流动相溶解并定容于100 mL容量瓶中,配制成8μg/mL的对照品贮备液。另称取黄体酮对照品2 mg,精密称定,用流动相溶解并定容,配制成200 ng/mL的黄体酮内标溶液。将贮备液置4℃冰箱保存,备用。
2.2 检测条件
色谱条件:色谱柱为 AcquityUPLCBEHC18柱(50mm×2.1mm,1.7 μm,Waters公司),柱温 40 ℃ ,流动相为乙腈 -含 0.01% 甲酸的水溶液(70 ∶30),流速 0.3mL /min,进样量 10 μL。
质谱条件:采用电喷雾正电离源(ESI+),多反应离子监测模式(MRM),毛细管电离电压3.18 kV,锥孔电压16 V,脱溶剂气温度600℃,气流量1 000 L/h,碰撞气体为 Ar气,碰撞能量20 V,源温度150℃。
2.3 样品预处理
取正常人群体检剩余血液样品,经抗凝剂EDTA-2K作用下提取血浆,作为本试验的模拟血浆样品。取模拟血浆样品1 mL,加入内标贮备液 35μL、pH=4.9的磷酸盐缓冲液 300μL、乙酸乙酯 3 mL,涡旋萃取 1.5 min,离心(4 000 r/min)3 min,取上层有机相于50℃水浴氮气挥干,残留物用500μL流动相涡旋溶解5min,离心(4 000 r/min)3min,经 0.22 μm 微孔滤膜过滤,进样10μL。
2.4 干扰试验
按2.2项下条件测定,质谱图见图1。阿托伐他汀与内标物质在各自离子通道上无内源性杂质干扰,色谱峰形良好,阿托伐他汀的保留时间约为0.63min,黄体酮的保留时间约为0.91min。
2.5 标准曲线与检测灵敏度试验
精密吸取阿托伐他汀对照品贮备液适量,以空白血浆为溶剂,配成质量浓度为 0.01,0.05,0.10,0.20,0.50,1.00,5.00,10.00,20.00,50.00 ng /mL 的系列含对照品的血浆样品。按 2.3项下方法处理后,以阿托伐他汀与内标物峰面积比值(R)对质量浓度(C)进行回归分析,得回归方程 R =0.160 7C+0.000 5,r=0.998 9(n=10)。另按同样方法配制 0.01 ng/mL 的标准含药血浆,按2.3项下方法处理后,用流动相反复稀释,依法测定,得最低检测质量浓度为 0.002 ng/mL(S /N≥3)。

图1 质谱图
2.6 萃取回收率试验
取空白血浆,加入适量的阿托伐他汀对照品贮备液,配成低、中、高(0.02,1.00,50.00 ng/mL)3 种质量浓度的标准含药血浆样品各5份。按2.3项下方法处理后分析,记录阿托伐他汀峰面积(A1);另取空白血浆 1 mL,按 2.3项下处理,水浴、氮气挥干后,残余物用 0.02,1.00,50.00 ng /mL 3 种质量浓度的对照品溶液溶解,进样,记录阿托伐他汀峰面积(A2)。以 A1/A2计算萃取回收率。结果见表1。
表1 阿托伐他汀萃取回收率试验结果(n=5,±s)

表1 阿托伐他汀萃取回收率试验结果(n=5,±s)
质量浓度(n g/mL)0.0 2 1.0 5 0.0萃取回收率(%)7 6.5 ± 2.6 7 7 5.0 ± 1.8 1 7 3.2 ± 0.9 4 R S D(%)3.4 9 2.4 1 1.2 9
2.7 回收率和精密度试验
取阿托伐他汀对照品溶液及空白血浆,配成低、中、高(0.02,1.00,50.00 ng/mL)3 种质量浓度的标准含药血浆样品,各 5 份,以2.3项下方法处理,进样分析,所得结果代入线性回归方程计算得实测质量浓度,并与已知加入质量浓度相比,得方法回收率。同法配制3种质量浓度含药血浆样品,依法处理,于同日内5次、连续5日进样分析,考察精密度。结果见表2,方法回收率为95% ~106%,日内、日间精密度的 RSD均≤5%。
表2 阿托伐他汀回收率和精密度试验结果(n=5,±s)

表2 阿托伐他汀回收率和精密度试验结果(n=5,±s)
质量浓度(n g /mL)回收率(%) 精密度的 R S D(%)0.0 2 1.0 5 0.0±s 9 8.0 ± 4.4 7 9 9.3 ± 2.7 8 1 0 3.0 ± 2.9 4 R S D 4.5 6 2.1 4 2.6 5日内4.6 1 1.5 4 2.4 0日间4.5 6 3.9 0 3.7 1
2.8 样品稳定性试验
取空白血浆,加入适量阿托伐他汀对照品贮备液,配成质量浓度分别为 0.02,1.00,50 ng /mL 的低、中、高含药血浆样品各两份,分别于常温或-20℃反复冻融,按2.3项下方法处理后进样测定,共测定5 d,以第1天测得质量浓度为100%,考察稳定性。结果见表3。
表3 阿托伐他汀稳定性试验结果(n=5,±s)
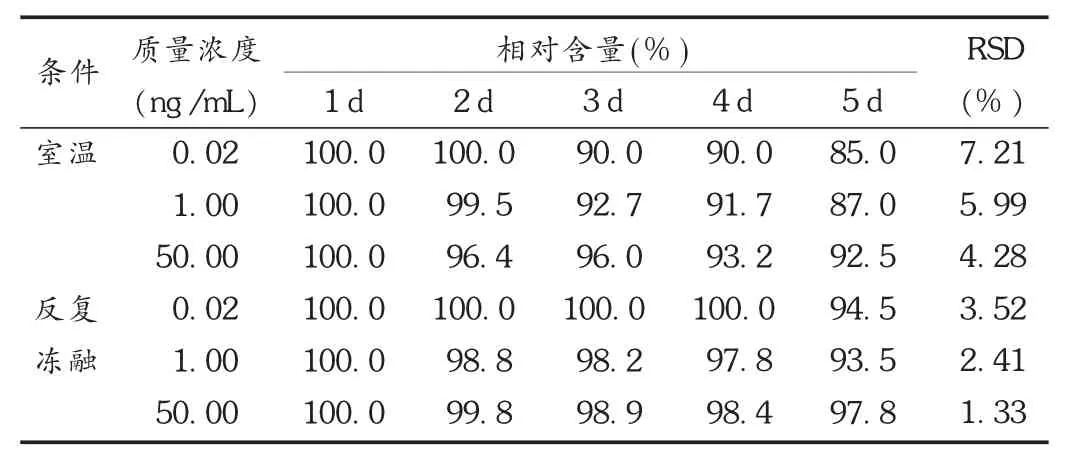
表3 阿托伐他汀稳定性试验结果(n=5,±s)
条件相对含量(%)室温反复冻融质量浓度(n g /mL)0.0 2 1.0 0 5 0.0 0 0.0 2 1.0 0 5 0.0 0 1 d 1 0 0.0 1 0 0.0 1 0 0.0 1 0 0.0 1 0 0.0 1 0 0.0 2 d 1 0 0.0 9 9.5 9 6.4 1 0 0.0 9 8.8 9 9.8 3 d 9 0.0 9 2.7 9 6.0 1 0 0.0 9 8.2 9 8.9 4 d 9 0.0 9 1.7 9 3.2 1 0 0.0 9 7.8 9 8.4 5 d 8 5.0 8 7.0 9 2.5 9 4.5 9 3.5 9 7.8 R S D(%)7.2 1 5.9 9 4.2 8 3.5 2 2.4 1 1.3 3
3 讨论
目前阿托伐他汀血药浓度的测定方法有高效液相色谱-紫外分光光度(HPLC-UV)法[3]和高效液相色谱 -质谱(HPLCMS)法[4]。前者灵敏度较低,无法较灵敏地检测出阿托伐他汀消除相中的血药浓度。后者报道的最低检出质量浓度为0.25 ng/mL,本试验对其作适当改进,使血药浓度最低定量限达0.01 ng/mL,保留时间由4.16min缩短到1min以内,提高了分析准确度、灵敏度和分析效率。
本试验中采用液-液萃取的方法处理血样。在萃取剂的选择中,比较了环己烷、二氯甲烷、氯仿和环己烷-二氯甲烷(3.5∶1)对阿托伐他汀的萃取效果,结果显示,这些试剂虽可减少杂质干扰,但萃取回收率均低于35%;以正戊烷为萃取剂时,提取回收率较高,但样品容易乳化。以乙酸乙酯萃取,可显著减少杂质干扰,且两次萃取的回收率可达90%,比仅萃取1次的回收率提高约20%,由于一次萃取已可使生物样品的纯化和浓集满足分析要求,操作更便捷,故采取一次萃取。
阿托伐他汀的相对分子质量为557.71,而用质谱测得的质荷比是559.2,可能是在离子化过程中阿托伐他汀获得1个质子后所得离子化物质的质荷比。同理,黄体酮的相对分子质量是314.47,而检测到的黄体酮质荷比是315.5。
选择流动相时,比较了乙腈-甲醇-水[5]和乙腈 -含0.01%甲酸的水溶液(70∶30),结果前者的阿托伐他汀色谱峰有拖尾现象,且阿托伐他汀在乙腈中的稳定性比甲醇好,因此用乙腈为溶剂和流动相的成分。乙腈含量过高时,阿托伐他汀保留时间过短,且易受血中杂质干扰,适当降低乙腈的含量至70%使阿托伐他汀的保留时间在0.62 min时,药物及内标可与血浆中内源性物质达到基线分离。流动相中加入适当的甲酸,可增加阿托伐他汀的离子化效应,显著提高方法的灵敏度和分离效果。
以内标法定量可改善试验的准确度和精密度。文献[6]采用尼莫地平为内标,考虑到有可能受临床伍用该药而影响测定,故选用黄体酮为内标。在拟订的色谱条件下,黄体硐可与待测物适当分离,与待测物和内源性杂质在各自的离子通道上不会互相干扰,且1 min内可完成测定。黄体酮为人体内源性物质,在正常人群中的含量均小于0.002 ng/mL,占本试验中所加入内标含量的0.04%,对测定影响较小。
文献[7]报道,阿托伐他汀在人体内的有效血药浓度为0.25~25 ng/mL,而本试验所测得的血药浓度为 0.01~50 ng/mL,这为临床研究阿托伐他汀消除相的浓度范围时提供了良好的依据。
综上所述,文中拟订的方法准确、灵敏、稳定、简便,适用于阿托伐他汀血药浓度测定。
参考文献:
[1]Loe AP,MeTaviish D.Atorvastatin.A review of its pharmacology and therapeutic potential in the management of hyperlidaemias[J].Drugs,1997,53(3):828.
[2]Malhotra HS,Cos KL.A Atorvastatin.An updated review of itspharmacologicalpropertiesand use in dyslipidaemia[J].Drugs,2001,61(12):1 835.
[3]Bahrami G,Mohammadi B,Mirzaeei S,et al.Determination at atorvastatin in human serum by reversed-phase high-perpormation liquid chromatography with UV detection[J].JChromatogr B Analyt Technol Biomed Life Sci,2005,826(1-2):41-45.
[4]Bullen WW,Miller RA,Hayes RN,et al.Development and validation of a high-perpormance liquid chromatography landem mass spectrometry assay for atorvastatin artho-hydroxy atorvastatin,para-hydroxy atorvastatin in human,dog,and rat plasma[J].J AM Soc Mass Spectrom,1999,10(1):55-56.
[5]Erk N,Altuntas TG.Liquid chromatographic determination of atorvastatin in bulk drug,tablets,and human plasma[J].JLiq Chromatogr Relat Technol,2004,27(1):83.
[6]刘 巍,张 庆.液相色谱-串联质谱法测定人血浆苯磺酸氨氯地平与阿托伐他汀钙浓度[J].医药导报,2010,29(6):698-700.
[7]William W Bullen,Ronald A Miller,Roger N Hayes.Development and Validation of a High-Performance Liquid Chromatography Tandem Mass Spectrometry Assay for Atorvastatin,Ortho-Hydroxy Atorvastatin,and Para-Hydroxy Atorvastatin in Human,Dog,and Rat Plasma[J].JAm Soc Mass Spectrom,1999,10:55-66.
猜你喜欢
煤化工(2022年3期)2022-07-08
浙江化工(2022年1期)2022-02-19
口腔护理用品工业(2021年4期)2021-11-02
合成纤维工业(2021年5期)2021-10-31
中华养生保健(2020年8期)2021-01-14
中华养生保健(2020年7期)2020-11-16
首都食品与医药(2020年1期)2020-10-21
中成药(2018年6期)2018-07-11
山东工业技术(2016年10期)2016-09-06
中国当代医药(2015年8期)2015-03-01
