
富氮多孔碳纤维的制备及其在超级电容器中的应用
2014-02-03 02:03张寿春吕春祥
化学工业与工程 2014年3期
李 莹,张寿春,吕春祥
(1.碳纤维制备技术国家工程实验室,中国科学院山西煤炭化学研究所,山西 太原030001;2.中国科学院大学,北京100049)
超级电容器又被称作电化学电容器或双电层电容器,具有比能量密度大、充电速度快、使用寿命长等优点,弥补了传统静电电容器和电池的不足,从而被广泛用作后备电源、替换电源和主电源。石墨烯、碳纳米管、活性碳、活性碳纤维等炭材料具备物理化学性能稳定、导电性高等优点,作为超级电容器电极材料表现出优异的性能[1-5]。其中,活性碳纤维由于具有高比表面积而具有更高的比电容。传统活性碳纤维的制备方法主要有活化法[6-7]和软/硬模板法[8],这些方法存在以下缺点:活化法对设备的腐蚀严重;硬模板法需要酸洗或碱洗去除模板剂,后处理工艺复杂;而软模板法的制备工艺复杂,尤其是嵌段共聚物的合成对聚合条件要求苛刻。此外,这些方法主要引入的是小/中孔,难以引入大/中孔。因此,探索活性碳纤维制备的新方法引起了学术界和产业界的重视。本实验以聚丙烯腈(PAN)为前驱体,在不需要模板和活化工艺的条件下,通过相分离调控制备具有丰富大/中孔结构的三维PAN纤维网络,采用水合肼和盐酸羟胺对PAN纤维进行预交联化处理,考察了化学交联作用对孔结构的影响及N原子对赝电容的贡献。
1 实验
1.1 原料
实验所需聚丙烯腈(PAN)由碳纤维制备技术国家工程实验室提供,聚四氟乙烯悬浮液(PTFE,质量分数为60%)、水合肼(分析纯)、盐酸羟胺(分析纯)均购买自上海晶纯生化科技股份有限公司。
1.2 富氮多孔碳纤维的制备
采用湿法纺丝制备PAN纤维,通过调节凝固浴中二甲基亚砜/水的配比,温度和拉伸比例制备三维PAN纤维网络,然后用去离子水连续清洗2 h去除PAN纤维网络中的二甲基亚砜溶剂,并室温干燥,得到具有丰富大/中孔的PAN纤维(PF)。将PF置于0.5 mol/L水合肼和0.5 mol/L盐酸羟胺混合溶液中进行预交联处理,于95℃恒温20 h再使用去离子水和乙醇依次超声清洗3次,60℃鼓风干燥箱干燥6 h,得到交联后的PAN纤维(LPF)。再将LPF于鼓风干燥箱中在190,220和250℃分别恒温2 h,使样品充分预氧化,接着将预氧化样品置于管式炉中,700℃N2氛围中恒温1 h,升温速率为10℃/min,制备的富氮多孔碳纤维(LCF)。选用普通不含大/中孔网络结构的PAN纤维(NPF)为对比样,并采用同样的工艺条件进行预氧化和碳化,制得碳纤维(NCF)。
1.3 材料的表征
采用JSM-7001F型场发射扫描电子显微镜(FE-SEM),JW-BK122W型氮气物理吸附仪对样品的形貌和孔结构进行表征,使用ESCALAB 250XI型X射线光电子能谱测定LCF的表面杂原子。
1.4 电极制备及电化学性能测试
将LCF和NCF纤维充分研磨,分别与乙炔黑、导电石墨和黏结剂PTFE按质量比75∶10∶10∶5混合均匀后涂覆在12 mm×12 mm的泡沫镍表面,置于鼓风干燥箱中60℃干燥4 h后,再置于真空干燥箱中60℃干燥12 h,得到的活性物质质量6~8 mg。
实验采用CHI660C电化学工作站,以6 mol/L KOH为电解液,使用三电极装置,采用汞/氧化汞电极作参比电极、铂片电极作对电极,测定材料的循环伏安曲线和恒电流充放电曲线。电极材料的比电容通过放电时间得到,计算公式为:

Cg为质量比电容,F/g;I为放电电流,A;ΔV为放电电压,V;Δt为放电时间,s;m为活性物质质量,g;Cs为比表面电容,F/m2;S为材料的比表面积m2/g。
2 结果与讨论
2.1 微观结构及表征
如图1所示,通过相分离调控得到的PF纤维具有发达的三维网络大/中孔结构,并且在采用水合肼和盐酸羟胺进行预交联反应后,LPF中的大/中孔结构基本保持不变,在进一步经预氧化和碳化后,LCF中的大/中孔结构虽然有所收缩,但大部分大/中孔得以保留。主要是因为大/中孔的骨架结构得到预交联和预氧化的双重巩固化作用。在预交联过程中,PAN纤维与水合肼和盐酸羟胺发生化学交联反应,其反应式如式(3)[9-10],交联结构使分子的稳定性增加。在随后的预氧化过程中,未交联的氰基发生热环化反应,形成耐热稳定的梯形结构,如式(4)[11]。水合肼、盐酸羟胺与PAN分子的化学交联反应和预氧化过程中氰基环化反应的共同作用抑制分子在热处理过程中的收缩,使PF中的大/中孔结构在LCF中大部分得以保留。而普通PAN纤维缺乏初始的大/中孔结构,经过预氧化的环化反应[式(4)]和碳化过程的热缩聚[式(5)][12]作用形成结构致密的片层石墨结构,如图1中的NPF和NCF所示。

图1 纤维横截面的FE-SEM照片Fig.1 FE-SEM images of cross sections of fibers
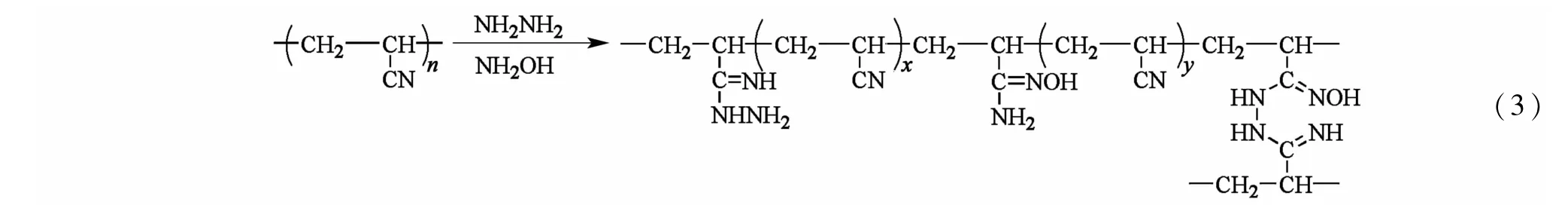
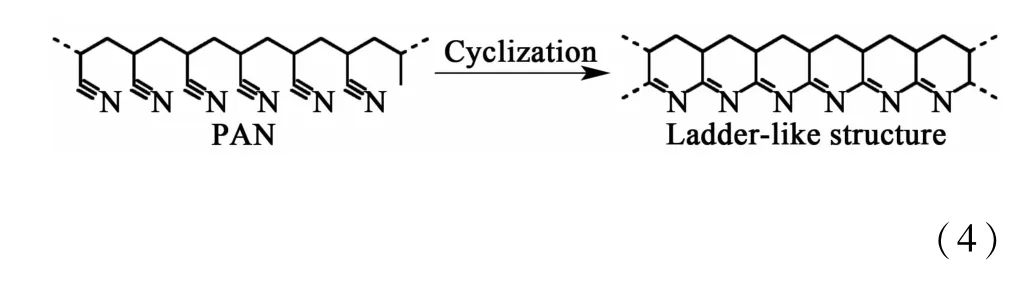
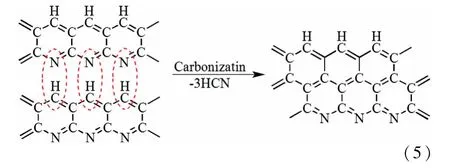
图2为吸附等温曲线,结果表明,PF和 LPF的吸附等温线主要表现为 IV型,说明 PF和 LPF是大/中孔材料;LCF的吸附等温线表现为 I和 IV型的复合形式,说明在碳化过程中有大量微孔产生,是一种多级孔结构材料;而NCF吸脱附等温线上的N2平衡吸附量较小,说明其孔含量较低。
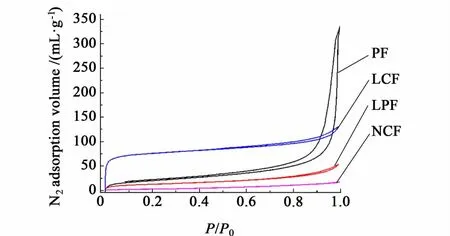
图2 吸脱附等温线Fig.2 N2 adsorption/desorption isotherm s of samples
表1为样品的比表面积和孔体积参数。其中,LPF转化为 LCF时,微孔显著增加,主要原因是PAN分子间与水合肼和盐酸羟胺高度交联[式(3)],一定程度上影响了预氧化过程中的环化反应,从而影响了碳化过程中的热缩聚反应,未进入梯形结构的交联键或者氰基在碳化过程热解产生微孔。而由NPF纤维制备的NCF,在经过预氧化和碳化后易形成片层石墨结构,结构致密,因热解产生的孔较少。

表1 纤维样品的比表面积及比体积Table 1 Specific sur face area and volume of fiber samples
2.2 电化学性能表征
如图3所示,通过循环伏安曲线测定了材料的电化学性能。图3a)中,LCF的CV曲线表现出类似理想电容器的矩形特征,而NCF的CV曲线更接近于三角形。图3b)中,在大扫描速率(50 mV/s)下,LCF仍然能保持类似矩形的特征,这主要与其孔道结构直接相关。LCF和NCF样品的CV曲线都有明显的变形,但无明显的氧化还原峰,与文献中报道的富氮碳材料在KOH电解液中的CV曲线形状相接近[13]。
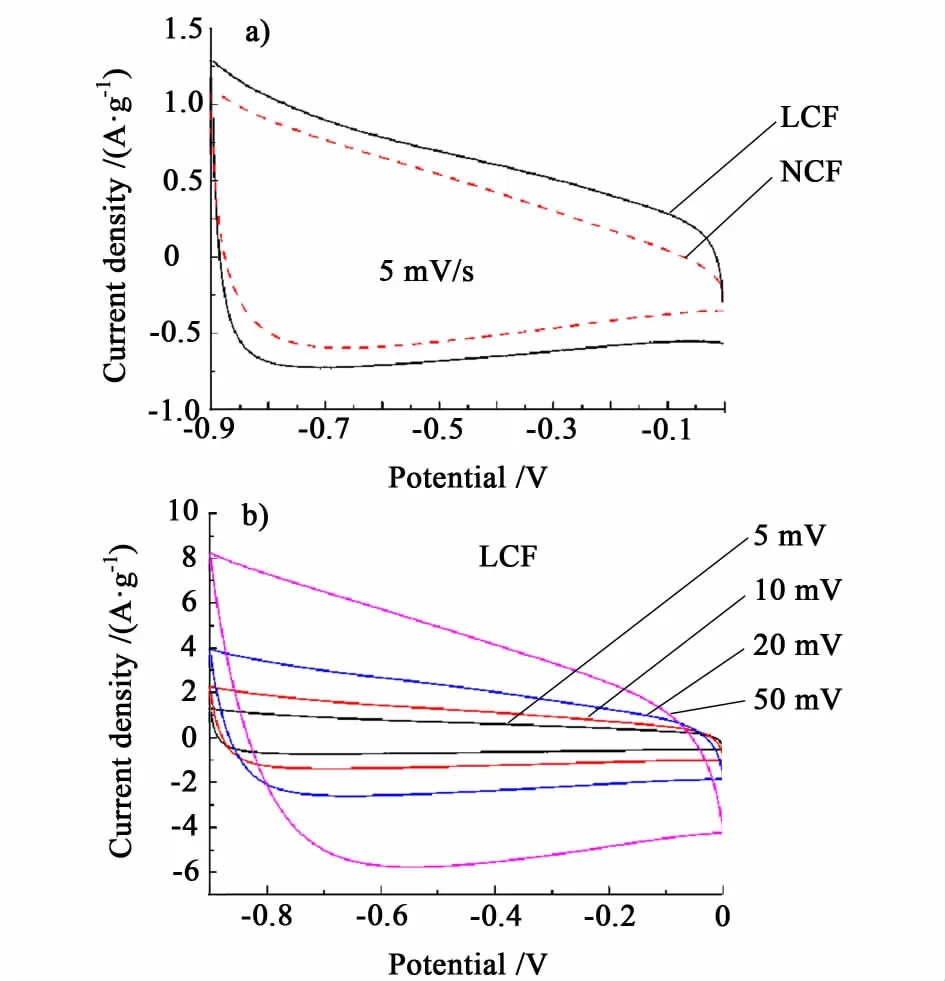
图3 a)扫描速率5 m V/s时LCF和NCF的循环伏安曲线;b)LCF在不同扫描速率下的循环伏安曲线Fig.3 a)CV cu rves of LCF and NCF at the scan rate of 5 m V/s;b)CV curves of LCF at different scan rates
为了验证杂原子对材料赝电容的贡献,采用XPS表征手段对LCF的表面杂原子进行了分析,如图4所示。图4a)证实LCF的材料表面存在大量的N、O杂原子,其原子含量分别是13.53%和8.01%。图4b)和图4c)中,对 N1s和 O1s的分析表明,N原子在材料中主要以吡啶型氮(N-6,398.7±0.3 eV)、吡咯型氮(N-5,400.3±0.3 eV)和 4价氮(N-Q,401.4±0.3 eV)的形式存在:O原子主要以醌型氧(OI,530.7 ±0.2 eV)、-C O(OII,531.9 ±0.2 eV)、C-OH/C-O-C(OIII,533.3 ±0.2 eV)和化学吸附氧或水(OIV,534.2 ±0.2 eV)的形式存在[6,14-15]。其中 N-6、N-5和 OI具有电化学活性[16],能够提供赝电容,其含量分别是4.6%、6.5%和2.1%;而 NQ只起导电作用,其含量为2.4%。由 N-5、N-6和OI的原子含量可见,N原子对赝电容的贡献较大,与CV曲线相吻合。
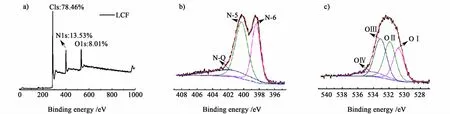
图4 LCF的a)全扫描,b)N1s高分辨,c)O1s高分辨XPS能谱Fig.4 XPS spectra of LCF:a)fu ll scan;high resolu tion b)N1s,c)O 1s
图5为样品在不同电流密度下的恒电流充放电曲线。LCF在电流密度为0.1 A/g和10.0 A/g时,比电容分别为222 F/g和110 F/g,电容保留率为50.0%,而NCF在电流密度为0.1 A/g和10.0 A/g时,比电容分别是142 F/g和23 F/g,电容保留率仅有16.2%。LCF和NCF样品的充放电曲线均不是完全对称的三角形,这是因为法拉第效应使放电曲线偏移,放电时间延长,电容量增加[参见公式(2)]。在大电流密度时,NCF的电容损失严重,主要是因为缺少有效的孔道结构,阻碍离子的传输。在LCF中,微孔结构贡献了双电层电容,而大/中孔结构起到电解质离子的缓存和传输作用,孔壁上大量的杂原子改善了电解液与材料的润湿性,同时增加了赝电容,提高材料的比表面利用率,使其比表面电容可达到0.80 F/m2。孔结构与杂原子的共同作用,使LCF的比电容与NCF相比提高了56.3%。

图5 电流密度为a)0.1 A/g,b)10.0 A/g时恒电流充放电曲线;c)LCF和NCF的比电容-电流密度关系曲线Fig.5 Charge-discharge curves at a curren t density of a)0.1 A/g and b)10 A/g;c)the specific capacitance of LCF and NCF at d ifferent curren t densities
3 结论
以PAN为前驱体制备了富氮多孔碳纤维,制备过程中采用水合肼和盐酸羟胺处理PAN纤维,不仅通过交联反应使孔骨架结构得到巩固,大/中孔结构得以保留的同时,产生更多微孔,并且在PAN链结构中进一步引入了氮原子。富氮多孔碳纤维中的N原子大部分以具有电化学活性的吡啶氮和吡咯氮形式存在,有利于提高赝电容,在0.1 A/g时电容为222 F/g,比表面电容达到0.80 F/m2。
[1]Jiang H, Lee P S, Li C.3D carbon based nanostructures for advanced supercapacitors[J].Energy&Environmental Science, 2013, 6(1):41_53
[2]Chen C,Zhang Q,Zhao X,et al.Hierarchically aminated graphene honeycombs for electrochemical capacitive energy storage[J].Journal of Materials Chemistry,2012,22(28):14 076_14 084
[3]Lota G,Lota K,Frackowiak E.Nanotubes based composites rich in nitrogen for supercapacitor application[J].Electrochemistry Communications, 2007, 9(7):1 828_1 832
[4]Chen X,Chen C,Zhang Z,et al.Gelatin-Derived nitrogen-doped porous carbon via a dual-template carbonization method for high performance supercapacitors[J].Journal of Materials Chemistry A, 2013, 1(36):10 903_10 911
[5]Chen L,Zhang X,Liang H,et al.Synthesis of nitrogen-doped porous carbon nanofibers as an efficient electrode material for supercapacitors[J].ACS Nano,2012,6(8):7 092_7 102
[6]Ma C,Song Y,Shi J,Zhang D,et al.Preparation and one-step activation of microporous carbon nanofibers for use as supercapacitor electrodes[J].Carbon, 2013,51:290_300
[7]Im JS,Jang JS,Lee Y S.Synthesis and characterization of mesoporous electrospun carbon fibers derived from silica template[J].Journal of Industrial and Engineering Chemistry, 2009, 15(6):914_918
[8]Im JS,Park S J,Lee Y S.Preparation and characteristics of electrospun activated carbon materials havingmeso-and macropores[J].Journal of Colloid and Interface Science, 2007, 314(1):32_37
[9]韩正邦,董永春,马斌,等.不同 PAN纤维铁配合物光催化偶氮染料降解反应[J].太阳能学报,2011,32(3):408_417 Han Zhenbang, Dong Yongchun, Ma Bin, et al.Degradation of azo dye catalized by different PAN fiber-Fe complexes[J].Acta Energiae Solaris Sinica, 2011, 32(3):408_417(in Chinese)
[10]赵振新,马步伟,梁志宏,等.硫代酰胺基螯合纤维的合成及其吸附性能研究[J].离子交换与吸附,2006,22(5):392_401 Zhao Zhenxin, Ma Buwei, Liang Zhihong, et al.Studies on synthesis and adsorption properties of thioamidegroup chelating fiber[J].Ion Exchange and Adsorption,2006,22(5):392_401(in Chinese)
[11]张利珍,吕春祥,吕永根,等.聚丙烯腈纤维在预氧化过程中的结构和热性能转变[J].新型碳材料,2005,20(2):144_152 Zhang Lizhen, Lu Chunxiang, Lu Yonggen, et al.The structure and thermal performance of PAN fibers during oxidative stabilization [J].New Carbon Materials,2005,20(2):144_152(in Chinese)
[12]Xue Y, Liu J, Lian F, et al.Effect of the oxygen-induced modification of polyacrylonitrile fibers during thermal-oxidative stabilization on the radial microcrystalline structure of the resulting carbon fibers[J].Polymer Degradation and Stability, 2013, 98(11):2 259_2 267
[13]Hulicova-Jurcakova D, Kodama M, Shiraishi S, et al.Nitrogen-Enriched nonporous carbon electrodes with extraordinary supercapacitance[J].Advanced Functional Materials, 2009, 19(11):1 800_1 809
[14]Biniak S, Szymański G, Siedlewski J, et al.The characterization of activated carbons with oxygen and nitrogen surface groups[J].Carbon, 1997, 35(12):1 799_1 810
[15]Arrigo R, Hävecker M, W rabetz S, et al.Tuning the acid/base properties of nanocarbons by functionalization via amination[J].Journal of the American Chemical Society, 2010, 132(28):9 616_9 630
[16]Yang X, Wu D, Chen X, et al.Nitrogen-Enriched nanocarbons with a 3-D continuous mesopore structure from polyacrylonitrile for supercapacitor application[J].The Journal of Physical Chemistry C,2010,114(18):8 581_8 586
猜你喜欢
氯碱工业(2022年6期)2022-11-21
北京大学学报(自然科学版)(2022年3期)2022-06-17
化工进展(2021年10期)2021-11-03
化工环保(2021年2期)2021-04-25
化工生产与技术(2021年6期)2021-02-18
云南化工(2020年5期)2020-06-12
盐科学与化工(2019年11期)2019-12-04
电子制作(2019年12期)2019-07-16
四川水力发电(2018年6期)2018-03-26
化学反应工程与工艺(2017年6期)2017-06-12
