
高效分子排阻色谱法测定头孢氨苄原料药中聚合物的含量
2013-12-03 03:35:48苑华张冬黄亚龙张文胜石家庄食品药品检验所石家庄05001河北省食品药品检验院石家庄050011华北制药河北华民药业有限责任公司石家庄050000
中国药房 2013年5期
苑华,张冬,黄亚龙,张文胜(1.石家庄食品药品检验所,石家庄05001;.河北省食品药品检验院,石家庄 050011;.华北制药河北华民药业有限责任公司,石家庄 050000)
研究[1]表明,β-内酰胺类抗菌药物引发的速发型过敏反应并非抗菌药物本身所致,而是与药物中存在的高分子聚合物杂质有关。头孢氨苄收载于2010年版《中国药典》(二部)[2],其质量标准中未对高分子聚合物进行控制。为此,笔者建立了以TSK-GEL G2000SWXL为色谱柱的高效分子排阻色谱法[3]分析测定头孢氨苄中聚合物含量的方法,结果表明该法分离效能高、检验周期短、结果准确可靠。
1 材料
LC-20AT高效液相色谱仪(日本岛津公司)。
头孢氨苄对照品(中国食品药品检定研究院,批号:130408-200710,纯度:94.4%);头孢氨苄原料药样品(华北制药河北华民药业有限责任公司,样品1:批号:20120101,纯度:95.1%;样品2:批号:20120102,纯度:95.2%;样品3:批号:20120103,纯度:95.2%);乙腈为色谱纯,其他试剂均为分析纯。
2 方法与结果
2.1 色谱条件
色谱柱:TSK-GEL G2000SWXL(300 mm×7.8 mm,5 μm);流动相:0.005 mol/L磷酸盐缓冲液[0.005 mol/LNaH2PO4溶液-0.005 mol/L Na2HPO4(39∶61,V/V)]-乙腈(95∶5,V/V),流速:0.6 ml/min;检测波长:220 nm;进样量:20 μl。
2.2 溶液的制备及测定方法
取样品适量,精密称定,加水制成每1 ml中含头孢氨苄2.0 mg的溶液,作为供试品溶液(临用新制);另取头孢氨苄对照品适量,精密称定,加水制成每1 ml中约含20µg的溶液,作为对照溶液。分别进样测定,按外标法以峰面积计算保留时间小于头孢氨苄的杂质总量。结果见图1。
2.3 破坏试验
高温破坏:取头孢氨苄对照品溶液(2.0 mg/ml)10 ml,于60℃水浴中放置10 min,取出,冷却至室温。
碱破坏:称取头孢氨苄对照品约20 mg,置于10 ml量瓶中,加0.1 mol/L NaOH溶液1.0 ml溶解,放置1 min后,加0.1 mol/L HCl 1.0 ml,用水稀释至刻度。
酸破坏:称取头孢氨苄对照品约20 mg,置于10 ml量瓶中,加0.1 mol/L HCl溶液1.0 ml溶解,放置4 h后,加0.1 mol/L NaOH 1.0 ml,用水稀释至刻度。
氧化破坏:称取头孢氨苄对照品约20 mg,置于10 ml量瓶中,加3%H2O2溶液1.0 ml溶解,放置5 min后,用水稀释至刻度。
光破坏:取头孢氨苄对照品溶液(2.0 mg/ml)10 ml,于5000 lx照度下放置8 h。
采用选定的色谱条件取上述溶液进样测定。结果表明,破坏处理之后的样品杂质峰均有所增加,且各主峰前聚合物峰均能与主成分峰充分分离,其保留时间与未破坏样品溶液色谱图中的聚合物峰一致。色谱见图1。
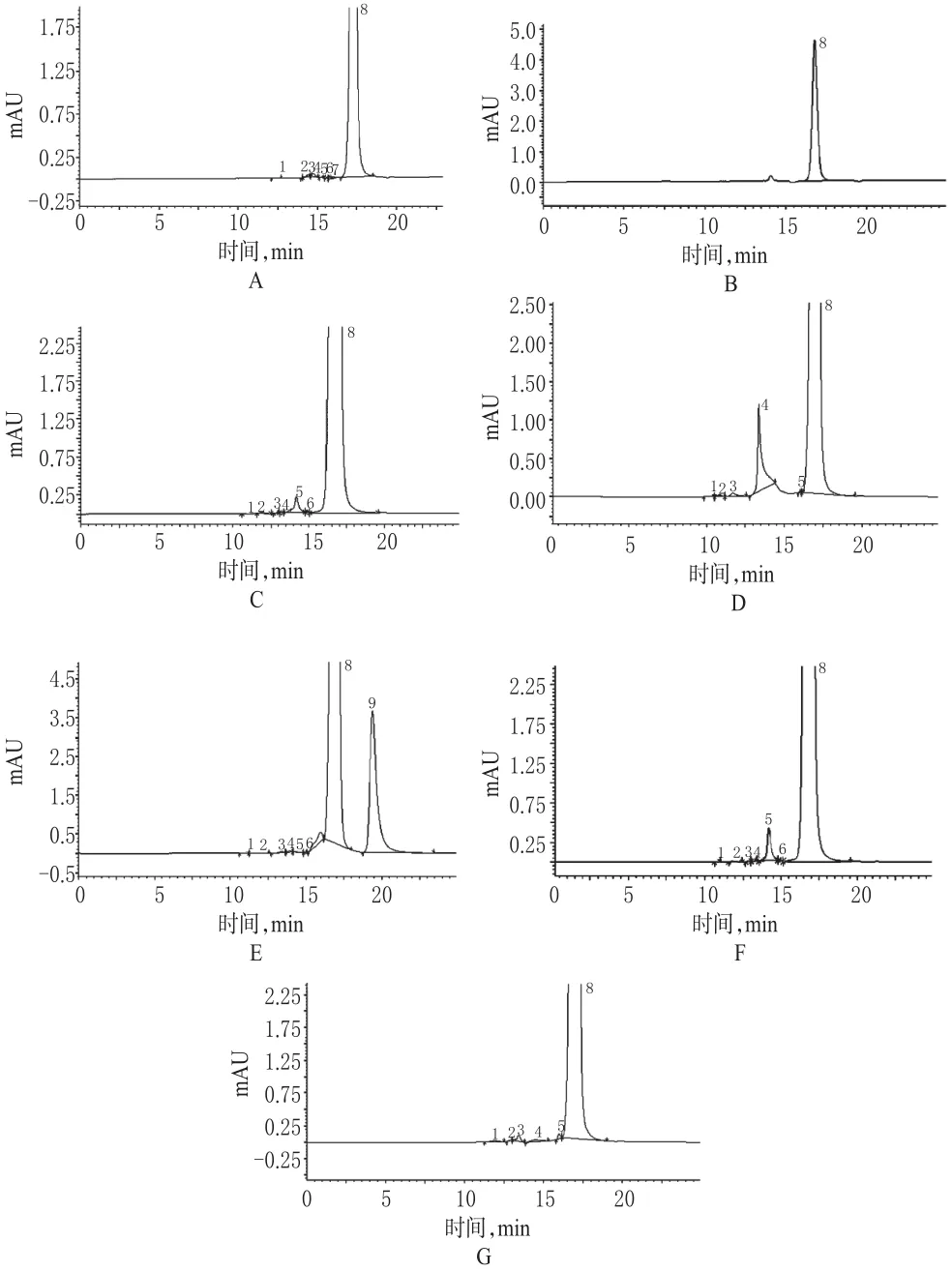
图1 高效液相色谱图Fig 1 HPLC chromatograms
2.4 标准曲线的建立
精密称取头孢氨苄对照品适量,加水溶解并稀释制成系列线性试验溶液,质量浓度分别为0.56、1.0、2.0、3.5、7.0、14.0、28.0 μg/ml,进样测定,并以相应组分的色谱峰面积(A)对其质量浓度(c)进行线性回归,得回归方程如下:A=55592c-1474.1(r=0.9999)。结果表明,头孢氨苄检测质量浓度线性范围为0.56~28.0 μg/ml。
2.5 检测限及定量限试验
精密称取头孢氨苄对照品适量,分别用水稀释成每1 ml中含头孢氨苄10、5、2.5、1、0.5 μg的系列溶液,进样测定,按信噪比为3测得头孢氨苄检测限为1.0 ng,按信噪比为10测得定量限为3.0 ng。
2.6 精密度试验
取质量浓度为7.0 μg/ml对照品溶液连续进样6次,结果头孢氨苄峰面积的RSD=0.3%(n=6),表明本法精密度好。
2.7 重复性试验
平行制备6份供试品溶液,分别立即进样分析,测定聚合物的总量。结果RSD=2.4%(n=6),表明本法重复性良好。
2.8 稳定性试验
取含头孢氨苄2.0 mg/ml的供试品溶液适量,在室温下放置,于0、1、2、3、4、5、6、7、8 h分别测定,8 h内杂质峰面积之和逐渐增大,3~4 h增加较大,表明供试品溶液不稳定。因此供试品溶液应临用新制。
2.9 样品中聚合物的测定
按“2.2”项下方法对3批样品进行检测,结果见表1。

表1 3批样品中聚合物测定结果Tab 1 Results of content determination of polymers in 3 batches of samples
3 讨论
(1)缓冲液的选择。在前期试验中笔者对不同离子强度的磷酸盐缓冲液作为流动相进行了考察,A为0.0025 mol/L,B为0.005 mol/L,C为0.01 mol/L。试验结果表明,A与C所检出的杂质峰较少,B所检出的杂质峰较多且实现了主峰与杂质峰间的有效分离,故选择0.005 mol/L磷酸盐缓冲液。
(2)流动相中乙腈比例的选择。分别选取乙腈比例为5%、10%进行考察。结果,乙腈比例为5%时检出色谱峰较多,故流动相选用乙腈比例为5%。
(3)流动相pH值的选择。头孢类抗菌药物在碱性、偏酸性及酸性条件下极易发生聚合反应,测得的聚合物含量难以反映样品本身的实际情况。故笔者参考2010年版《中国药典》(二部)相关内容,采用pH值7.0的磷酸盐缓冲液为流动相。
(4)检测波长的选择。用二极管阵列检测器测定碱破坏供试品溶液,采集到主峰前各高分子杂质峰的紫外光谱图,杂质1~3的最大吸收波长集中于220~230 nm波长范围内;杂质4未见明显吸收峰,但于240~200 nm波长范围内呈现递增的吸收状态;杂质5于262 nm波长处呈现最大吸收,其吸收值与220 nm波长处的吸收值较接近;头孢氨苄于257 nm波长处呈现最大吸收,其吸收值亦与220 nm波长处的吸收值较接近。为保证最大杂质检出量,检测波长定为220 nm。
(5)由于结构不同的聚合物通常具有相同的生物学特性,且聚合物又具有高度的不均一性,因此在药品质量控制中只需控制聚合物总量即可[1]。
(6)本方法与以葡聚糖凝胶Sephadex G-10作为分离介质的分子排阻法[4]相比较,具有检验周期短(本文方法约20 min,葡聚糖凝胶Sephadex G-10方法约为60 min)、操作简单、分离效果好的优点,这可能是因为TSK-GEL G2000SWXL系球型硅胶基质在其表面通过共价键化学键合亲水基团构成的物质,其吸附及分子筛效应强于Sephadex G-10葡聚糖凝胶颗粒。以TSK-GEL G2000SWXL为色谱柱的方法是凝胶色谱发展的必然趋势。
[1]袁雯玮.高分子聚合物研究与中国药典2005年版B2内酰胺类抗生素高分子聚合物修订情况及操作要点[J].中国抗生素杂志,2005,30(12):727.
[2]国家药典委员会.中华人民共和国药典:二部[S].2010年版.北京:中国医药科技出版社,2010:208.
[3]王成刚,曹轶,张喆,等.高效分子排阻色谱法分析头孢噻肟钠中的聚合物[J].药物分析杂志,2009,29(5):814.
[4]伍蓉.分子排阻色谱法测定头孢氨苄聚合物[J].国外医药抗生素分册,2012,33(2):83.
猜你喜欢
中国科技纵横(2021年24期)2021-03-02 06:42:52
艺术品鉴(2020年6期)2020-12-06 10:49:08
家庭医学(下半月)(2020年7期)2020-04-18 13:45:31
中成药(2018年6期)2018-07-11 03:01:12
领导文萃(2017年6期)2017-03-24 09:31:39
北方牧业(2016年1期)2016-12-17 19:08:50
中学生数理化·高一版(2016年7期)2016-12-07 20:47:07
中学生数理化·中考版(2015年12期)2015-09-10 07:22:44
中国医药生物技术(2014年4期)2014-01-23 09:24:24
山西大同大学学报(自然科学版)(2013年5期)2013-09-13 10:44:14
