
海蚬多糖性质及抗氧化活性研究
2013-11-09 00:44蒋长兴焦云鹏熊清平陈晓明王纪忠曾晓雄
食品与生物技术学报 2013年10期
蒋长兴, 焦云鹏, 熊清平, 陈晓明, 王纪忠, 曾晓雄
(1.淮阴工学院 生命科学与化学工程学院,江苏 淮安 223003;2.南京农业大学 食品科技学院,江苏 南京210095;3.江苏食品职业技术学院,江苏 淮安 223003)
多糖是生物体内广泛存在的一类大分子物质,一些天然多糖已被证实具有抗肿瘤、抗病毒、抗感染、抗氧化、增强免疫力等多种生物学功能[1]。多糖的这些生物学功能与其自身的理化性质、空间结构、取代基的类型等方面密切相关[2]。较多研究表明,海蚬多糖具有抗肿瘤、增强免疫力、抗感染等生理功能[3]。然而到目前为止有关海蚬多糖的分离纯化、理化性质及活性等方面的研究报道不多。采用离子交换柱层析 (DEAE-纤维素)与凝胶柱层析(Sephadex G-100)法,将海蚬多糖进行分离纯化,获得不同纯化组分;测定粗多糖及其纯化组分的总糖质量分数、蛋白质质量分数、糖醛酸质量分数、硫酸基质量分数,以及相对分子质量及纯度,测定粗多糖及纯化产品的还原力、金属离子螯合能力、还原能力、脂质过氧化抑制活性等,对海蚬多糖及纯化组分的抗氧化活性进行体外评价。
1 材料与方法
1.1 材料与试剂
新鲜海蚬:南京惠民桥水产品批发市场提供;DEAE-纤维素、葡聚糖G-100、葡萄糖醛酸、葡聚糖标准品:购于Sigma公司;考马斯亮蓝G-250、氯化钡、明胶、三氯乙酸、三氟乙酸等:均为分析纯试剂。
1.2 仪器与设备
可见分光光度计:721型,上海菁华科技仪器有限公司产品;电子天平:AY-120型、BL-220H型,日本SHIMADZU公司产品;电脑全自动部份收集器:DBS-100A型,上海沪西分析仪器厂产品;气相色谱仪:Agilent 6890N型,美国Agilent公司产品;高效液相色谱仪:Agilent 1100 Series型,美国Agilent公司产品;旋转蒸发器:Laborota-400型,Heidolph公司产品。
1.3 方法
1.3.1 提取制备 鲜活海蚬清洗干净,暂养过夜。去壳,取肉、体积分数75%乙醇保存。2 d后将肉匀浆,向匀浆中加入体积分数75%乙醇保存4周,以脱去海蚬匀浆中的脂肪、色素及其它杂物。离心,烘干,得海蚬组织粉。称取海蚬组织粉适量,加入一定体积的蒸馏水,水浴提取、离心、抽滤、浓缩、体积分数75%乙醇沉淀、烘干得多糖粗提物。Sevag法脱蛋白,得粗多糖,备用。
1.3.2 分离纯化 海蚬多糖DEAE-纤维素层析柱的分离与葡聚糖凝胶柱层析法进一步纯化参照Qiao等[4]报道的方法并稍作修改。称取60 mg海蚬多糖溶于3 mL去离子水中,加样于DEAE-纤维素层析柱(2.6 cm×30 cm)中,用不同浓度的NaCl溶液依次洗脱,洗脱液流速设为1.0 mL/min,自动部分收集器分步收集(10 min/管)不同浓度洗脱液对应的流出液。硫酸-苯酚法检测各管流出液的多糖含量(490 nm处吸收值),将较大吸光值对应的流出液合并收集,合并液50℃条件下旋转蒸发浓缩,,透析24 h,经真空冷冻干燥,获得初步纯化产品。称取20 mg经DEAE-纤维素初步纯化的多糖组分,溶于5 mL去离子水中,加样于Sephadex G-100凝胶层析柱(2.6 cm×60 cm)。用去离子水洗脱,洗脱速度为0.25 mL/min,自动部分收集器分步收集流出液(5 min/管)。硫酸-苯酚法检测各管中流出液的多糖含量(490 nm处吸收值),将较大吸光值对应的流出液合并收集,合并液于50℃条件下旋转蒸发浓缩,自来水和去离子水分别透析24 h,最后将透析液真空冷冻干燥,得到纯化产品。
1.3.3 理化性质测定 样品中总糖、蛋白质、糖醛酸硫酸基质量分数的测定,参照Dubois等[5]、Bradford[6]、Blumenkrantz 等[7]、Dodgson 等[8]等 报 道 的方法。样品的纯度和相对分子质量测定,参照Roy等[9]报道的分子排阻高效液相色谱法。将已知相对分子质量的标准多糖用流动相配成1.0 mg/mL标准溶液,经0.45 μm滤膜过滤后,采用Agilent 1100 series高效液相色谱仪、TSK-Gel G3000 SWXL色谱柱、示差检测器(RID)进行分析。流动相为含0.1 mol/L Na2SO4的0.01 mol/L磷酸盐缓冲液(pH 6.8),流动相流速为0.8 mL/min,色谱柱和检测器温度均为25℃,进样体积为20 μL,记录示差色谱图的保留时间(TR)。绘制多糖分子量的标准曲线。称取多糖样品配制1.0 mg/mL溶液,相同条件下进行高效液相色谱分析。
1.3.4 体外抗氧化活性
1)金属螯合能力的测定 金属离子螯合能力的测定参照Liu等[10]报道的方法。取不同浓度的样品(CP、F1、F2和 F3)溶液 1.0 mL 于试管中,分别加入0.05 mL氯化亚铁溶液(2.0 mmol/L)、0.2 mL菲洛嗪(ferrozine)溶液(5.0 mmol/L)、2.75 mL 去离子水。 混匀反应体系,10 min后分光光度法测定562 nm处吸光值。设EDTA-2Na为实验对照。金属离子螯合能力按照下面公式进行计算:螯合率(%)=(A对照-(A样品-A干扰))/A对照×100%。
2)还原力的测定 还原力测定参照Oyaizu等[11]报道的方法。配制不同浓度的待测试样1.0 mL,分别加入1.0 mL磷酸盐缓冲液(0.2 mol/L,pH=6.6)和1.0 mL 铁氰化钾[K3Fe(CN)6]溶液(1%)。50 ℃水浴20 min,迅速冷却,加入三氯乙酸(质量分数10%)1.0 mL,离心(5 000 r/min,15 min)。 取上清液 1.5 mL,依次加入 1.5 mL H2O,0.3 mL FeCl3溶液(质量分数0.1%),混匀,700 nm处测定吸光值,得A1。以0.3 mL H2O代替FeCl3溶液(质量分数0.1%),测得A2。设Vc为实验对照。还原力为A700nm=(A样品-A干扰)。
3)脂质过氧化抑制活性的测定 脂质过氧化抑制活性的测定采用硫代巴比妥酸反应底物(TBARS)法[12]。取不同浓度的多糖溶液 1.0 mL,依次加入1.0 mL的质量分数1%的小鼠肝脏匀浆 (每100 mL的匀浆含有1 g的小鼠肝脏)、0.05 mL的0.5 mmol/L FeCl2溶液、0.05 mL的 0.5 mmol/L H2O2溶液,混合物37℃反应60 min后,再加入1.5 mL的TCA溶液(质量分数20%)、1.5 mL TBA溶液(质量分数0.8%),所得混合液100℃反应 15 min,4 000 r/min离心10 min后,分光光度法测定上清液在532 nm处的吸光值,设Vc为实验对照。抑制率(%)= (A对照-(A样品-A干扰))/A对照×100%。
2 结果与分析
2.1 海蚬多糖分离纯化
采用DEAE-纤维素柱层析法,以不同浓度NaCl溶液(0、0.1、0.5 mol/L)作为洗脱液对海蚬粗多糖进行初步分离,得到3个主要组分,分别标记为F1、F2、F3(图 1)。 将这 3 个组分分别收集,50 ℃减压蒸发浓缩,浓缩液去离子水透析48 h,透析液真空冷冻干燥,得到初步纯化产品。利用Sephadex G-100层析柱对经DEAE-纤维素色谱柱初步分离的3个组分进行纯化处理,得到3个纯化组分(图1)。分别记为 f1、f2和 f3。
2.2 海蚬多糖基本性质

图1 海蚬多糖DE-52(a)与葡聚糖G-100(b)柱层析洗脱曲线Fig.1 Stepwise elution curve of CP on anion-exchange chromatography column of DEAE-52 cellulose(A)and elution curve of polysaccharide fractions(F1,F2and F3)from DEAE-52 cellulose on sizeexclusion chromatography column of Sephadex G-100 (B)
海蚬多糖基本化学组成测定显示:海蚬粗多糖总糖、蛋白质质量分数分别为83.81%、3.08%。表明海蚬经过热水提、醇沉等工艺所得的产品多糖为主要成分。由于海蚬多糖属于动物性多糖,含有一定量的糖蛋白,采用Sevag法只能除去游离蛋白质,因此海蚬粗多糖的蛋白质可能是结合的糖蛋白。海蚬多糖纯化组分f1、f2、f3总糖质量分数分别为98.75%、95.58%、84.71%,f3蛋白质质量分数为 6.34%,f1、f2蛋白质未能检出。表明f1、f2为单一的不含蛋白的多糖组分,而f3含有一定量结合的糖蛋白。海蚬粗多糖通过DEAE-纤维素与Sephdex G-100凝胶柱层析法分离纯化所得到的各个组分总糖质量分数、纯度均大大提高,并且能够将含有结合蛋白的组分与其他多糖组分进行有效地分离。
由糖醛酸、硫酸基质量分数测定结果可以看出,海蚬多糖糖醛酸质量分数分别为1.58%、0.16%、0.96%、2.13%,硫酸基质量分数分别为0.92%、1.22%、2.08%、3.58%。表明海蚬多糖均含有不等量的糖醛酸、硫酸基等带电基团,随着NaCl浓度的提高,所得到的纯化组分糖醛酸、硫酸基含量逐渐提高。该结论证实f2、f3为含有糖醛酸、硫酸基的酸性多糖组分。f1含有一定量的糖醛酸与硫酸基,与f2、f3相比,含量均较低,因此可以认为f1是一种中性多糖。
2.3 海蚬多糖的纯度及相对分子质量
海蚬多糖分子排阻高效液相图谱表明f1、f2、f33组分在高效液相图谱上的保留时间分别为8.423、8.140、7.750 min,将保留时间代入回归方程,计算得到各组分的平均相对分子质量。f1、f2、f3的平均相对分子质量分别为 68 600、80 600、100 600。
2.4 体外抗氧化活性
2.4.1 金属离子的螯合能力 海蚬多糖对金属离子螯合能力的测定结果见图2。由图2可以看出,海蚬多糖具有一定金属离子的螯合能力。多糖浓度对金属离子的螯合能力影响极显著(P<0.01)。随着多糖质量浓度提高,金属离子的螯合能力呈上升趋势。对CP来说,当多糖质量浓度在0~3.2 mg/mL的范围内时,金属离子的螯合能力随着多糖质量浓度的提高呈较显著的上升趋势;当多糖质量浓度在3.2~4.0 mg/mL的范围内时,金属离子的螯合能力随着多糖质量浓度的提高上升趋势不显著 (P>0.05)。对 f3、f2、f1来说, 在实验设计的范围内 (0~4.0 mg/mL),金属离子的螯合能力随着多糖质量浓度的提高呈较显著的上升趋势。在实验设计的范围内(0~4.0 mg/mL),海蚬多糖的金属离子的螯合能力由小到大的顺序为 F1<F2<F3<CP。 多糖质量浓度为 4.0 mg/mL 时,CP、F1、F2、F3的金属离子的螯合能力分别为95.1%、57.7%、63.5%、86.3%。由以上分析知,海蚬多糖高质量浓度时对金属离子的螯合能力较强。有报道称,化合物中如果含有-OH、-SH、-COOH、-PO3H2、C=、-NH2、-S-、-O-等基团就有可能具有一定的金属离子螯合能力[13]。海蚬多糖中的对金属离子的螯合能力可能是由于其分子链中含有的大量羟基、羧基等活性基团。
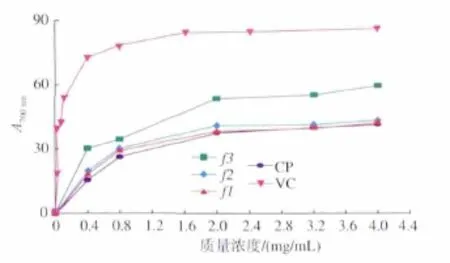
图2 海蚬粗多糖及纯化组分的金属离子螯合能力Fig.2 Chelating effects and reducing power of CP,F1,F2 and F3
2.4.2 海蚬多糖的还原力 海蚬多糖还原力的测试结果见图3。由图3可以看出,海蚬多糖具有一定还原能力。多糖浓度对还原力影响达到显著水平(P<0.05)。随着多糖质量浓度提高,还原力呈上升趋势。在实验设计的范围内(0~4.0 mg/mL),海蚬多糖的还原力由小到大的顺序为 CP<F1<F2<F3。 多糖质量浓度为 4.0 mg/mL 时,CP、F1、F2、F3还原力分别为41.6%、42.9%、43.4%、59.6%。在实验设计的范围内(0~4.0 mg/mL),海蚬多糖的还原力均显著低于Vc,表明海蚬多糖具有中等强度的还原力。
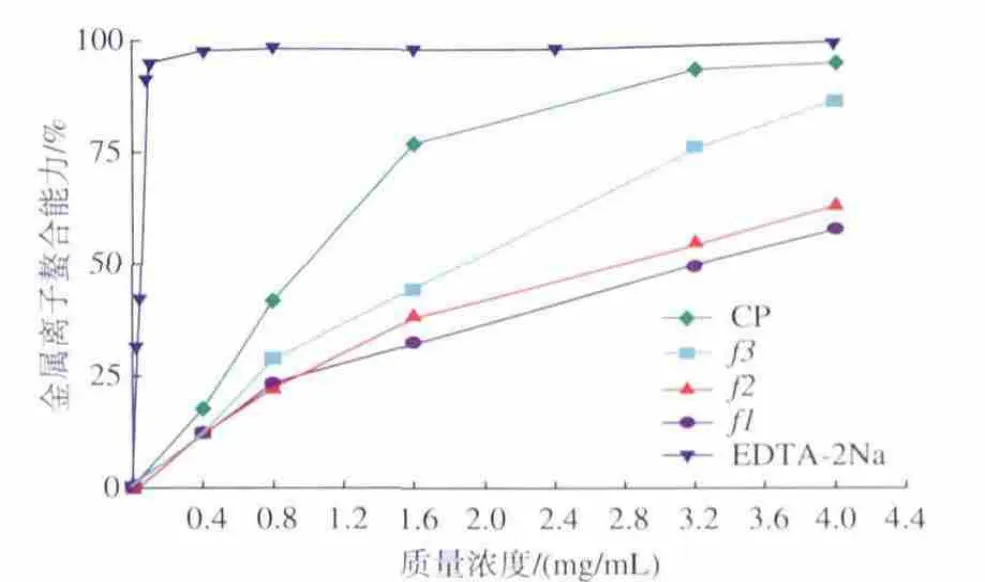
图3 海蚬粗多糖及纯化组分的金属离子螯合能力Fig.3 Chelating effects and reducing power of CP,F1,F2 and F3
2.4.3 海蚬多糖的脂质过氧化抑制活性 脂质过氧化是生物体内氧自由基参与的不饱和脂肪酸自动氧化反应。生物体内脂质过氧化反应一方面破坏了细胞膜结构的完整性,另一方面生成了具有潜在的细胞毒性脂质过氧化产物。脂质过氧化产物通常是丙二醛(MDA),该物质的生成量是衡量生物体内脂质过氧化反应程度的重要指标[14]。MDA能与硫代巴比妥酸发生化学反应,生成一种粉红色物质,该物质在波长532 nm处呈最大吸收。在多糖的脂质过氧化抑制活性测试中,通过采用分光光度法测定反应体系532 nm处吸光值的变化来确定多糖脂质过氧化抑制活性的高低[15]。海蚬多糖脂质过氧化抑制活性的测试结果见图4。
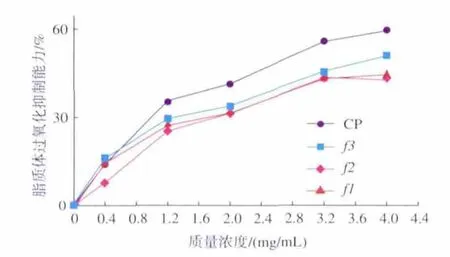
图4 海蚬粗多糖及纯化组分的脂质过氧化抑制活性Fig.4 Lipid peroxidation inhibition activity of CP,F1,F2 and F3
由图4可以看出,海蚬多糖具有一定脂质过氧化抑制活性。多糖质量浓度对脂质过氧化抑制活性影响达到极显著水平(P<0.01)。随着多糖质量浓度提高,脂质过氧化抑制活性呈上升趋势。对CP、F1、F2、F3来说,多糖质量浓度较低时(0~0.8 mg/mL)时,脂质过氧化抑制活性随着多糖质量浓度的提高呈较显著的上升趋势;多糖质量浓度较高时(3.2~4.0 mg/mL)时,脂质过氧化抑制活性随着多糖质量浓度的提高上升趋势不显著。
在实验设计的范围内(0~4.0 mg/mL),海蚬多糖的脂质过氧化抑制活性由小到大的顺序为F2<F1<F3<CP。 多糖质量浓度为 4.0 mg/mL 时,CP、F1、F2、F3的脂质过氧化抑制活性分别为59.6%、45.1%、43.4%、51.0%。高质量浓度时(3.2、4.0 mg/mL),海蚬多糖的脂质过氧化抑制能力与Vc较为接近,表明海蚬多糖具有具中等的脂质过氧化抑制活性。海蚬多糖的脂质过氧化抑制活性可能是其在清除自由基、螯合金属离子、还原能力等方面活性的综合反映。
3 结语
1)采用DEAE-纤维素柱层析法对海蚬多糖进行初 步 分离 , 获 得 3 个 组 分 (F1、F2、F3)。 采 用Sephdex G-100凝胶柱层析进一步分离纯化,获得3个纯化组分(f1、f2、f3)。 对 f1、f2、f3的纯度进行鉴定,结果表明,f1、f2、f3为均一的多糖组分。
2)采用苯酚-硫酸法、硫酸-间羟联苯法、考马斯亮蓝法、氯化钡-明胶法分别对海蚬多糖中总糖、糖醛酸、蛋白质、硫酸基含量进行分析。结果表明,CP、f1、f2、f3总糖质量分数分别为 83.81%、98.75%、95.58%、84.71%;糖醛酸质量分数分别为1.58%、0.16%、0.96%、2.13%;硫酸基质量分数分别为0.92%、1.22%、2.08%、3.58%;CP、f3蛋白质分别为3.08%、6.34%,f1、f2均未能检出;HPLC 分析结果表明,f1、f2、f3的平均相对分子质量分别为 68 600、80 600、100 600。
3)采用化学方法对 CP、f1、f2、f3进行体外抗氧化活性测定。结果表明,海蚬多糖具有不同的还原力、脂质过氧化抑制活性、金属离子螯合能力。
[1]Lu Y,Wang D,Hu Y,et al.Sulfated modification of epimedium polysaccharide and effects of the modifiers on cellular infectivity of IBDV[J].Carbohydrate Polymers,2008,71(2):180-186.
[2]Alban S,Schauerte A,Franz G.Anticoagulant sulfated polysaccharides:Part I.Synthesis and structure-activity relationships of new pullulan sulfates[J].Carbohydrate Polymers,2002,47(3):267-276.
[3]湛孝东.贝类多糖生物学活性研究进展[J].时珍国医国药,2006,17(7):1285-1286.ZHAN Xiao-dong.Progress on biological activities of polysaccharides from shellfish[J].LiShiZhen Medicine and Materia Medica Research,2006,17(7):1285-1286.(in Chinese)
[4]乔德亮,曾晓雄.三角帆蚌多糖制备及基本理化性质[J].食品与生物技术学报,2011,1:70-77.QIAO De-liang,ZENG Xiao-xiong.Preparation and preliminary feature of polysaccharides from Hyriopsis cumingii[J].Journal of Food Science and Biotechnology,2011,1:70-77.(in Chinese)
[5]Dubois M,Gilles K A,Hamilton J K,et al.Colorimetric method for determination of sugars and related substances[J].Analytical Chemistry,1956,28(3):350-356.
[6]Bradford M M.A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding[J].Analytical Biochemistry,1976,72(1-2):248-254.
[7]Blumenkrantz N,Asboe-Hansen G.New method for quantitative determination of uronic acids[J].Analytical Biochemistry,1973,54(2):484-489.
[8]Dodgson K,Price R.A note on the determination of the ester sulphate content of sulphated polysaccharides[J].Biochemical Journal,1962,84(1):106-110.
[9]Roy SK,Maiti D,Mondal S.Structural analysis of a polysaccharide isolated from the aqueous extract of an edible mushroom,Pleurotus sajor-caju,cultivar Black Japan[J].Carbohydrate Research,2008,343(6):1108-1113.
[10]Liu C,Wang C,Xu Z,et al.Isolation,chemical characterization and antioxidant activities of two polysaccharides from the gel and the skin of Aloe barbadensis Miller irrigated with sea water[J].Process Biochemistry,2007,42(6):961-970.
[11]Oyaizu M.Studies on products of the browning reaction.Antioxidative activities of browning reaction products prepared from glucosamine[J].Japanese Journal of Nutrition,1986,44(6):307-315.
[12]Yen G C,Hsieh C L.Antioxidant activity of extracts from Du-zhong (Eucommia ulmoides)toward various lipid peroxidation models in vitro[J].Journal of Agricultural and Food Chemistry,1998,46(10):3952-3957.
[13]Yuan Y V,Bone D E,Carrington M F.Antioxidant activity of dulse (Palmaria palmata)extract evaluated in vitro[J].Food Chemistry,2005,91(3):485-494.
[14]Janero D R.Malondialdehyde and thiobarbituric acid-reactivity as diagnostic indices of lipid peroxidation and peroxidative tissue injury[J].Free Radical Biology and Medicine,1990,9(6):515-540.
[15]Tsuda T,Watanabe M,Ohshima K,et al.Antioxidative activity of the anthocyanin pigments cyanidin 3-O-.beta.-D-glucoside and cyanidin[J].Journal of Agricultural and Food Chemistry,1994,42(11):2407-2410.
猜你喜欢
食品工业科技(2023年4期)2023-02-14
现代临床医学(2021年6期)2021-11-20
山东科学(2020年3期)2020-06-11
中国食品学报(2019年10期)2019-11-12
中成药(2018年9期)2018-10-09
中成药(2018年1期)2018-02-02
中成药(2017年12期)2018-01-19
中成药(2017年4期)2017-05-17
中国洗涤用品工业(2015年4期)2015-02-28
中国洗涤用品工业(2015年2期)2015-02-28
