
氨咖黄敏胶囊中2种组分质量控制方法的建立及9厂家产品质量评价
2013-11-09 08:33丁苏苏王威王玉李倚云闻琍毓江苏省扬州药品检验所江苏扬州225009中国药科大学南京20009江苏省药品检验所南京20008
中国药房 2013年5期
丁苏苏,王威,王玉,李倚云,闻琍毓(.江苏省扬州药品检验所,江苏扬州225009;2.中国药科大学,南京 20009;3.江苏省药品检验所,南京 20008)
氨咖黄敏胶囊,原名速效伤风胶囊,为临床常用的抗感冒药。其为复方制剂,每粒含对乙酰氨基酚250 mg、咖啡因15 mg、马来酸氯苯那敏1 mg、人工牛黄10 mg。
目前全国共有554家企业生产本品,现行标准[1]中采用滴定法分别测定其中对乙酰氨基酚和咖啡因的含量,测定方法较为烦琐、专属性差。另质量标准中亦无溶出度检查项,对处方中药物的溶出过程未能实施监控。为满足全面控制药品质量的要求,笔者采用高效液相色谱(HPLC)法同时测定该制剂中对乙酰氨基酚和咖啡因的含量,并在此基础上建立了溶出度检查方法,考察了9个厂家生产的该制剂的质量情况。
1 材料
1.1 仪器
LC-20AB HPLC仪,包括SPD-20A紫外检测器、LC Solution色谱工作站(日本岛津公司);RCZ-8M溶出试验仪,包括RZQ-8D自动取样收集系统(天大天发科技有限公司);AB135-S电子分析天平(瑞士梅特勒-托利多仪器有限公司)。
1.2 试剂与药品
对乙酰氨基酚(批号:100018-200408,纯度:100%)、咖啡因对照品(批号:171215-20809,纯度:99.9%)来源于中国食品药品检定研究院;氨咖黄敏胶囊(原研厂为广州白云山光华制药股份有限公司,批号:C11206,均为江苏省2011年评价性抽样品种,其余产品来自9个厂家,编号为A~I);乙腈为色谱纯,水为纯化水,其他试剂均为分析纯。
2 方法与结果
2.1 含量测定[2-4]
2.1.1 色谱条件。色谱柱:菲罗门Gemini-C18(250 mm×4.6 mm,5 μm);流动相:磷酸盐缓冲液(磷酸二氢铵11.5 g,加水溶解后,加H3PO41 ml,加水至1 000 ml)-乙腈(82∶18,V/V),流速:1.0 ml/min;柱温:30 ℃;检测波长:262 nm;进样量:20 μl。
取“2.1.3~2.1.5”项下3种溶液进样分析,结果各组分的分离度符合要求,色谱见图1。
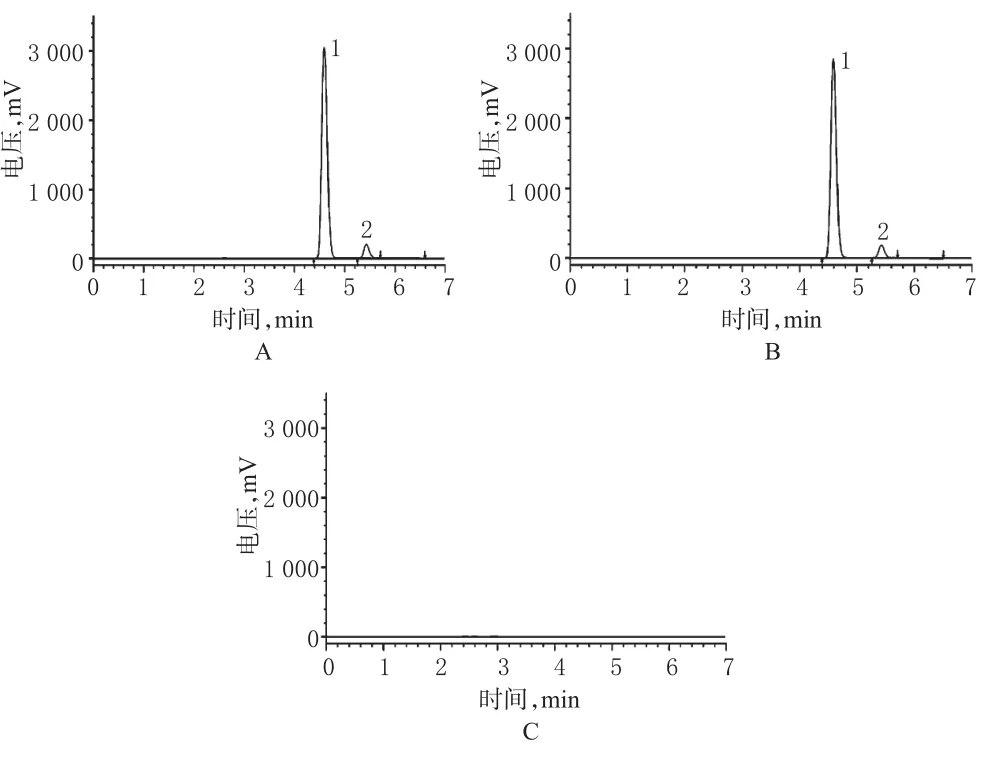
图1 高效液相色谱图A.对照品;B.供试品;C.阴性对照;1.对乙酰氨基酚;2.咖啡因Fig1 HPLC chromatogramsA.substance control;B.test sample;C.negative control;1.acetaminophen;2.caffeine
2.1.2 对照品贮备液的制备。分别精密称取对乙酰氨基酚、咖啡因对照品250.40、15.83 mg,置于100 ml量瓶中,加流动相溶解并稀释至刻度,即得。
2.1.3 对照品溶液的制备。精密称取对乙酰氨基酚对照品约15 mg,置于50 ml量瓶中,加流动相适量,超声使溶解;另取咖啡因对照品约18 mg,置于100 ml量瓶中,加流动相溶解并稀释至刻度,摇匀,精密量取5 ml至上述含有对乙酰氨基酚的量瓶中,加流动相稀释至刻度,即得。
2.1.4 供试品溶液的制备。取胶囊20粒的内容物,混匀,精密称取适量(约相当于对乙酰氨基酚125 mg),置于50 ml量瓶中,加流动相适量,超声处理20 min,加流动相稀释至刻度,摇匀,滤过;取续滤液3 ml,置于25 ml量瓶中,加流动相稀释至刻度,摇匀,即得。
2.1.5 阴性对照溶液的制备。按处方工艺,取降对乙酰氨基酚、咖啡因以外的其他药物制成阴性对照样品,按“2.1.4”项下供试品溶液的制备方法操作,即得。
2.1.6 线性关系考察。(1)对乙酰氨基酚。分别精密量取“2.1.2”项下对照品贮备液1.0、1.5、2.0、2.5、3.0、3.5、4.0、4.5、5.0 ml,置于25 ml量瓶中,用流动相溶解并稀释至刻度。分别进样分析,记录色谱图,以峰面积(A×106)对其质量浓度(c)进行线性回归,得回归方程为A=40.537c+0.938 6(r=0.999 9),结果表明对乙酰氨基酚检测质量浓度线性范围为0.100 2~0.500 8 mg/ml。(2)咖啡因。分别精密量取“2.1.2”项下对照品贮备液1、2、3、4、5、6、7、8、9、10 ml,置于25 ml量瓶中,用流动相溶解并稀释至刻度。分别进样分析,记录色谱图,以咖啡因的峰面积(A×105)对其质量浓度(c)进行线性回归,得回归方程为A=0.417 9c+0.012 6(r=0.999 9),结果表明咖啡因检测质量浓度线性范围为6.332~63.320 μg/ml。
2.1.7 精密度试验。取同一批号样品,制备供试品溶液1份,连续进样6次。结果,对乙酰氨基酚色谱峰峰面积的RSD=0.13%(n=6),咖啡因RSD=0.06%(n=6)。
2.1.8 重复性试验。取同一批号样品,制备供试品溶液6份,进样测定。结果,对乙酰氨基酚平均含量为99.90%,RSD=0.9%(n=6);咖啡因平均含量为93.23%,RSD=0.3%(n=6)。表明方法重复性较好。
2.1.9 稳定性试验。取同一批号样品,制备供试品溶液1份,室温放置,分别于0、2、4、6、8 h进样分析。结果,对乙酰氨基酚峰面积的RSD=0.5%(n=5);咖啡因峰面积的RSD=0.4%(n=5)。表明供试品溶液室温放置8 h稳定。
2.1.10 加样回收率试验。称取供试品9份,每份约0.285 g,精密称定,置于200 ml量瓶中,精密称取对乙酰氨基酚对照品约200、250、300 mg各3份,咖啡因对照品约12、15、18 mg各3份,分别加入上述9个200 ml量瓶中,加流动相适量,超声处理20 min,取出,冷却至室温,加流动相稀释至刻度,摇匀,滤过。取续滤液5 ml,置于25 ml量瓶中,用流动相稀释至刻度,摇匀。进样分析,记录色谱图,计算回收率。结果,对乙酰氨基酚平均回收率为100.81%,RSD=1.4%(n=9);咖啡因平均回收率为101.10%,RSD=0.7%(n=9)。
2.1.11 样品含量测定。取9个厂家的样品依法制备成供试品溶液,进样,记录峰面积;按外标法计算2种组分含量,结果见表1。

表1 9厂家样品含量测定结果Tab1 Content determination of samples from 9 manufacturers
2.1.12 含量测定结果分析。本文涉及样品均为江苏省2011年评价性抽样样品,笔者分别采用滴定法和本文建立的HPLC法对95批样品进行了含量测定,并用统计学方法对数据进行了统计。通过总体方差分析得出:对乙酰氨基酚的F=0.863 521,P=0.353 864,P>0.05;咖啡因的F=0.010 971,P=0.916 683,P>0.05。表明2种含量测定方法统计结果差异无显著性意义,详见表2。
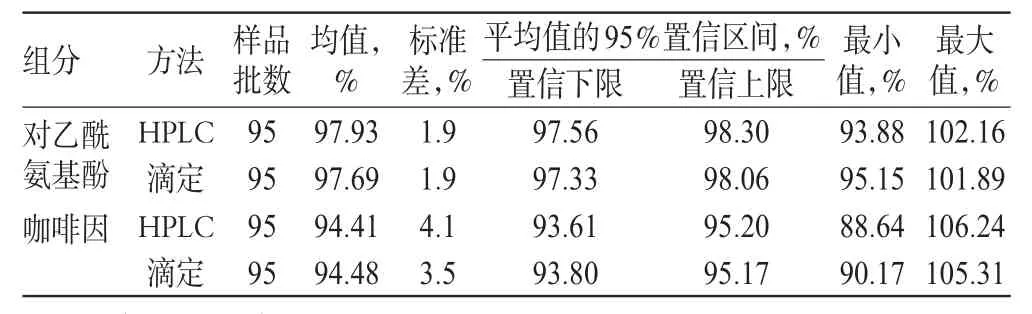
表2 2种含量测定方法结果统计分析Tab2 Statistics analysis of 2 kinds of methods
对HPLC法含量测定的结果进行统计分析,结果对乙酰氨基酚含量的频数分布图呈正态分布,咖啡因含量的频数分布图呈偏态分布。不同厂家样品中的对乙酰氨基酚含量的方差分析结果:F=6.394 2,P=1.82×10-8,P<0.05;咖啡因含量的方差分析结果:F=47.215 0,P=9.26×10-31,P<0.05。可认为各生产厂家的样品中2种组分含量测定结果具有显著性差异。
95批样品中,对乙酰氨基酚含量的平均值为97.93%,其中3.9%的样品含量在93.0%~94.9%之间,96.1%的样品含量在95.0%~102.9%之间;咖啡因含量的平均值为94.41%,其中15%的样品含量在88.0%~89.9%之间(均为A厂生产),49%的样品含量在90.0%~94.9%之间,只有36%的样品含量在95.0%~101.9%之间。分析咖啡因含量偏低的主要原因可能是一些厂家存在低限投料的情况。
2.2 溶出度试验[5]
2.2.1 溶出方法。采用篮法:转速为100 r/min,介质体积900 ml,温度为(37±0.5)℃,取样后照“2.1”项下方法测定。
2.2.2 溶出介质的选择。选择原研厂生产的1批样品,从中随机抽取4份,每份6粒,分别在水、盐酸溶液(0.1 mol/L)、磷酸盐缓冲液(pH 6.8)和醋酸盐缓冲液(pH 4.0)4种介质中,按“2.2.1”项下方法试验,分别在5、10、15、20、25、30、40 min定点取样5 ml,过滤,同时补充等量同温度的介质,进样测定,按外标法分别计算累积溶出百分率,绘制溶出曲线,见图2。

图2 4种溶出介质中2种组分的溶出曲线A.对乙酰氨基酚;B.咖啡因Fig2 Dissolution curves of 2 components in 4 dissolution mediumsA.acetaminophen;B.caffeine
根据图2结果,并参考《中国药典》中胶囊剂[3]的溶出介质,最后选择水作为溶出介质。
2.2.3 溶出曲线的比较。以水为溶出介质,对9厂家的样品进行体外溶出试验,分别计算在5、10、15、20、25、30、40 min时2种组分的累积溶出百分率,绘制溶出曲线,见图3。

图3 9厂家样品溶出曲线A.对乙酰氨基酚;B.咖啡因Fig3 Dissolution curves of samples from 9 manufacturersA.acetaminophen;B.caffeine
图3 结果表明,各厂样品中对乙酰氨基酚在15 min时累积溶出百分率均达到80%以上,20 min时均达到85%以上。除A厂外,其他各厂样品中咖啡因累积溶出百分率在15 min时均达到85%以上,A厂样品中咖啡因在25 min时达到80%。
2.2.4 相似因子(f2)的比较。以原研厂的样品为参比,计算各厂间溶出曲线f2值。f2值大于50则认为相似,结果见表3、表4。
从表3、表4结果可以看出,对乙酰氨基酚和咖啡因的f2值低于50者分别占60%和56%,说明各产品溶出曲线存在差异。
3 讨论
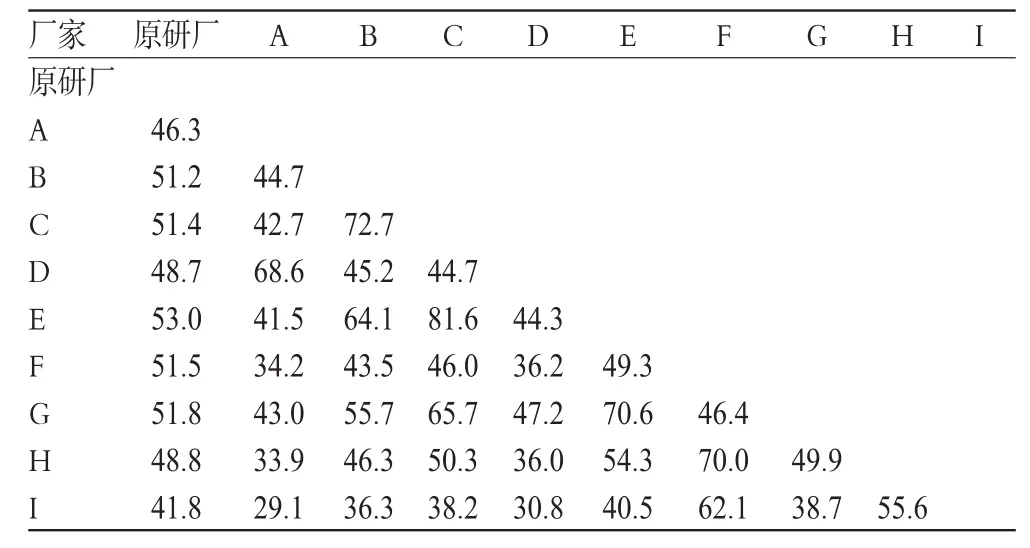
表3 对乙酰氨基酚的f2值比较Tab3 Comparison of f2of acetaminophen

表4 咖啡因的f2值比较Tab4 Comparison of f2of caffeine
预试验时,笔者分别对样品中2种组分进行紫外吸收波长扫描。结果发现,对乙酰氨基酚约为10 μg/ml时,在212、243 nm波长左右均有合适的吸光度;咖啡因约为10 μg/ml时,在220、262 nm波长左右均有合适的吸光度。由于该样品每粒含对乙酰氨基酚250 mg、咖啡因15 mg,二者的量相差很大,故吸收波长选择在咖啡因吸收较大处;再根据扫描的结果,初步确定2个检测波长:216、262 nm。试验发现,在216 nm波长处,2种组分的吸光度均稍大于262 nm波长处,但由于216 nm波长处溶剂峰的影响较大,最终确定以262 nm为检测波长。
比较该制剂2种组分含量测定的2种方法,虽然二者测定结果在统计学上无差异,但当采用滴定法测定时,A厂样品含量均≥90.0%;而采用HPLC法测定时,约15%样品中的咖啡因含量在88.0%~89.9%之间。因此对于检出厂家低限投料样品的不合格率,HPLC法更具优势。
从溶出度曲线和f2值比较的结果看,不同厂家氨咖黄敏胶囊的溶出存在差异,有必要在标准中增加溶出度检查项目,以全面控制该制剂的质量。
[1]国家食品药品监督管理局.WS-10001-(HD-0276)-2002-2006[S].2006.
[2]李爱华,韩香玲.RP-HPLC法测定复方氨酚烷胺胶囊中3种组分的含量[J].中国药品标准,2009,10(3):208.
[3]王小虹,修培,修锐.HPLC法同时测定氨咖黄敏胶囊2组分的含量[J].中国药事,2008,22(5):407.
[4]盖轲,郑阿利.高效液相色谱法同时测定氨咖黄敏胶囊中对乙酰氨基酚和咖啡因的含量[J].中国药房,2006,17(6):456.
[5]王威,李倚云,凌真,等.盐酸洛美沙星分散片体外溶出度测定方法的建立[J].中国药房,2012,23(13):1 218.
猜你喜欢
中国药学药品知识仓库(2022年13期)2022-07-03
药品评价(2021年17期)2021-11-06
杭州化工(2020年2期)2020-08-31
化工管理(2020年18期)2020-07-15
保健与生活(2020年4期)2020-03-02
海外星云 (2019年14期)2019-08-12
天然产物研究与开发(2018年5期)2018-06-13
大自然探索(2017年10期)2017-10-28
大自然探索(2017年5期)2017-05-26
中国妇幼健康研究(2017年3期)2017-01-15
