
Gab2 在SHP-2酪氨酸磷酸酶激活突变所致小鼠髓系异常增殖中的作用*
2013-10-24 06:22汪心怡李菲菲瞿成奎汪思应
中国病理生理杂志 2013年6期
陈 吉, 汪心怡, 陈 卓, 李菲菲, 郑 红, 瞿成奎,2, 汪思应
(1安徽医科大学病理生理教研室,安徽 合肥 230032; 2Case Western Reserve University, Cleveland, Ohio 44106 USA)
Gab2 在SHP-2酪氨酸磷酸酶激活突变所致小鼠髓系异常增殖中的作用*
陈 吉1, 汪心怡1, 陈 卓1, 李菲菲1, 郑 红1, 瞿成奎1,2, 汪思应1
(1安徽医科大学病理生理教研室,安徽 合肥 230032;2Case Western Reserve University, Cleveland, Ohio 44106 USA)
目的观察SHP-2信号通路中关键接头蛋白Gab2对SHP-2激活突变引发的小鼠髓系异常增殖是否具有调控作用。方法用Gab2-/-和SHP-2D61G/+模型小鼠建立4种基因型(SHP-2+/+、Gab2-/-、SHP-2D61G/+和SHP-2D61G/+/Gab2-/-)小鼠,解剖分析其脾大小,外周血白细胞计数,流式细胞术检测外周血及骨髓髓系细胞表面标志分子Mac-1和Gr-1并计数Mac-1和Gr-1阳性髓系细胞比例,骨髓造血干/祖细胞集落形成实验检测小鼠造血干细胞或祖细胞对细胞因子反应性,Western blotting和免疫沉淀实验检测骨髓来源肥大细胞经IL-3刺激后磷酸化蛋白激酶B (p-Akt)和磷酸化胞外信号调节激酶(p-ERK)的活化水平,以及Gab2与SHP-2蛋白的结合情况。结果敲除Gab2后显著减轻SHP-2激活突变导致的小鼠髓系增殖表型,主要表现在:脾指数减小,外周血白细胞减少,小鼠髓系来源的Mac-1和Gr-1阳性细胞比例降低。与SHP-2D61G/+小鼠相比,经IL-3刺激后,骨髓细胞的集落形成能力显著降低;骨髓来源肥大细胞内p-ERK和p-Akt表达明显下调,SHP-2D61G/+/Gab2-/-小鼠肥大细胞内无Gab2与SHP-2结合。结论敲除Gab2可以明显减轻SHP-2D61G/+激活突变导致的小鼠髓系异常增殖,这种减轻作用可能与SHP-2无法与Gab2结合而导致下游信号途径ERK和Akt活化减弱有关。
SHP-2酪氨酸磷酸酶; 基因激活突变; Gab2; 髓系增殖性疾病
髓系增殖性疾病(myeloproliferative disorders, MPD)属骨髓增生异常综合征(myelodysplastic syndrome, MDS)范畴,系干细胞来源的克隆性疾病。形态学观察可见病态造血现象,其特点是骨髓有核细胞增多,增殖的细胞可向终末分化成熟,多不伴发育异常;外周血红细胞、白细胞、血小板增多,可伴有肝脾肿大,后期出现骨髓纤维化、骨髓衰竭及转化为急性白血病。最终因MDS期骨髓衰竭伴随并发症或到达白血病期而难以治疗。为此MDS 也被称为白血病前期[1]。在修订的2008年WHO分类系统中,MPD改称为骨髓增殖肿瘤(myeloproliferative neoplasms, MPN),并强调了这类疾病的本质是肿瘤[1-3]。
MPD的发病机制不清,研究认为,蛋白酪氨酸激酶(protein tyrosine kinases,PTKs)信号转导途径过度活化是MPN重要的发病机制。同时也发现,PTKs信号通路负性调控基因PTPN11激活突变可能与其有关[4]。PTPN11编码的蛋白SHP-2是蛋白酪氨酸磷酸酶(protein tyrosine phosphatases, PTPs)家族成员之一,广泛表达于机体各组织细胞中,参与调节包括 Ras-ERK 在内的多条信号通路[5-7],与细胞的存活、迁移、黏附、细胞骨架形成等关系密切[8-9]。作为一种酪氨酸磷酸酶,SHP-2通过将与其结合的信号转导分子去磷酸化,从而阻断局部信号转导。但是,SHP-2在大部分信号通路中却是起到增强信号转导的作用。研究发现,SHP-2在造血细胞中高表达,对造血细胞的发育、分化起正调控作用,促进血细胞的增殖和分化[6,9]。此外,SHP-2在IL-3介导的造血细胞信号转导中除了起磷酸酶催化活性依赖的作用外,其还发挥了催化活性非依赖的接头蛋白功能[9-10]。2001年以来,SHP-2激活突变体在Noonan综合征(伴MPD)、MPD、幼年型粒单细胞白血病及其它白血病和一些实体瘤中陆续发现[11-14],提示SHP-2与白血病的发生密切相关。为了更好地研究SHP-2突变体引发疾病的机制,Neel实验室建立了SHP-2D61G/+基因敲入小鼠模型,在该模型小鼠中并没有发现白血病,但出现Noonan综合症和MPD表型[6,15-16]。
本课题组前期运用SHP-2D61G/+模型小鼠,观察SHP-2激活突变对小鼠髓系增殖及相关信号通路的影响,发现SHP-2D61G/+小鼠出现明显的髓系异常增殖,并且可能与Ras-ERK及 PI3K-Akt的活化有关[17]。进一步研究发现,SHP-2D61G/+小鼠分离得到的肥大细胞对IL-3反应性增高,可能由于在此过程中SHP-2发挥接头蛋白的功能,激活突变的SHP-2与Gab2结合增多所致[6,17-18]。
Gab2蛋白是SHP-2介导的信号通路中关键的接头蛋白,它属于Gab蛋白家族的一员,其结构中缺乏酶活性结构域,可以通过其自身的PH结构域、脯氨酸富集区及多个酪氨酸位点与其它信号分子结合,参与多种细胞信号调控,在细胞增殖、分化、凋亡及迁移等生理过程中发挥重要作用。目前认为,Gab2激活后主要通过下游的 Ras-ERK及 PI3K-Akt 等途径传递信号[19]。Gab2 结构中包含2个 SHP-2的绑定位点,是SHP-2重要的结合分子[18-21]。
由此设想,在SHP-2激活突变导致的髓系异常增殖过程中,减少Gab2的作用是否可以降低MPD的发生。为此,我们运用SHP-2D61G/+及Gab2-/-模型小鼠,建立SHP-2+/+、Gab2-/-、SHP-2D61G/+和SHP-2D61G/+/Gab2-/-4种基因型小鼠,探寻敲除Gab2对SHP-2激活突变导致的髓系异常增殖有无影响及其机制。
材 料 和 方 法
1材料
1.1药品与试剂 生理盐水购于北京双鹤药业,RPMI-1640培养基和胎牛血清购于Gibco,FITC标记的Mac-1和Gr-1抗体购于北京博奥森生物公司,Western blotting 所用ERK、Akt、p-ERK、p-Akt、SHP-2、p-Tyr、Gab2等抗体及 protein A/G琼脂糖珠均购于Santa Cruz。
1.2动物 C57BL/6品系SHP-2D61G/+及Gab2-/-小鼠由美国Case Western Reserve大学瞿成奎教授提供。用Gab2-/-和SHP-2D61G/+小鼠杂交,鉴定出雌雄SHP-2D61G/+/Gab2+/-小鼠作为种鼠杂交,生产并鉴定出本实验所需的4种基因型小鼠,分别为:SHP-2+/+、Gab2-/-、SHP-2D61G/+和SHP-2D61G/+/Gab2-/-。取10周龄小鼠配对备用。
2方法
2.1脾指数的测定 小鼠颈椎脱臼处死,称取体重(g),分离脾脏称重(mg),计算脾指数=脾重(mg)/体重(g)。
2.2外周血白细胞(white blood cell ,WBC)计数 小鼠摘取眼球取血约300 μL于2% EDTA-K2抗凝剂的采血管中,红细胞破坏法(3%乙酸溶液∶血液=19∶1)获得白细胞,应用F2800微细胞仪计数小鼠白细胞。
2.3骨髓细胞的分离 处死小鼠,无菌分离双侧股骨和胫骨并剪去骨端,用3 mL RPMI-1640培养液冲洗骨髓3次,收集冲洗液,吹打混匀成单细胞悬液,用白细胞稀释液稀释,作骨髓有核细胞计数后备用。
2.4流式细胞术检测骨髓细胞表面标记 骨髓细胞离心洗涤后,用含2% BSA的PBS制备成细胞悬液,加2 ng Fc受体阻断抗体,室温孵育15 min,离心洗涤1次,每管加入细胞1×106个,体积为100 μL,分别加FITC标记的相关抗体(Mac-1和Gr-1),冰浴30 min,加3 mL预冷PBS后,离心洗涤,每管加500 μL PBS(1% BSA)重悬细胞,流式细胞仪检测分析。
2.5骨髓细胞集落形成检测 骨髓细胞建立后,按2×107/L接种于含造血细胞生长因子IL-3的甲基纤维素半固体培养基中,37 ℃、5% CO2及饱和湿度的培养箱内进行培养。培养7 d后在倒置显微镜下观察集落形成,并分别计数骨髓细胞的集落数(≥50个细胞组成的细胞团)。
2.6骨髓中单个核细胞的分离 用来分离骨髓单个核细胞的分层液比重是1.120±0.001的聚蔗糖-泛影葡胺分层液。离心后,红细胞、粒细胞比重大,沉于管底;淋巴细胞和单核细胞的比重小于分层液比重,漂浮于分层液的上方,也可有少部分细胞悬浮于分层液中。吸取分层液液面的细胞,就可以从骨髓中分离到单个核细胞,主要包括淋巴细胞和单核细胞。将细胞置于另一短管中,加入5倍体积的Hanks液,离心洗涤细胞2次,弃上清,加入含有10% FBS和0.5 μg/L IL-3的RPMI-1640培养液,重悬细胞,常规培养。
2.7肥大细胞的获取和培养 骨髓细胞分离后,用含10% FBS和0.5 μg/L IL-3的RPMI-1640培养液,细胞按1.0×109/L接种于培养皿,37 ℃、5% CO2及饱和湿度的培养箱内进行培养,48 h后贴壁生长细胞为巨噬细胞,吸取悬浮细胞于另一培养皿中。每72 h更换培养基,每周更换培养皿,同样条件下培养2周后即为肥大细胞。
2.8Western blotting检测信号分子ERK及Akt的活化情况 取4组指数生长期的肥大细胞,饥饿培养6 h后,用2.0 μg/L IL-3刺激0、5和15 min,提总蛋白,10% SDS-PAGE凝胶电泳,转膜,封闭,分别加入p-ERK、p-Akt、Akt和ERK抗体 (4 ℃孵育过夜)及相应经辣根过氧化物酶标记的Ⅱ抗 (常温孵育1 h),用ECL系统进行检测。
2.9免疫沉淀(immunoprecipitation,IP)检测Gab2与SHP-2的结合情况 取4组指数生长期的肥大细胞,饥饿培养6 h后,用2.0 μg/L IL-3刺激0、5和15 min,预冷PBS洗涤,加入 IP裂解液缓冲液裂解1 h,离心取上清,加入protein A/G agarose,翻转混匀预吸附,离心取上清。按实验需要加入不同Ⅰ抗(SHP-2、Gab2和p-Tyr),翻转混匀,最后加入protein A/G agarose 20 μL,4 ℃翻转孵育过夜。洗涤,沉淀后加上样缓冲液,100 ℃,煮沸10 min。离心取上清,20 μL上样于15% SDS-PAGE进行Western blotting检测。
3统计学处理
数据用均数±标准差(mean±SD)表示,SPSS 13.0统计软件对数据进行方差分析。以P<0.05为差异有统计学意义。
结 果
1Gab2敲除后,由SHP-2突变导致的小鼠脾脏增大表型明显改善
与SHP-2+/+小鼠相比,Gab2敲除小鼠脾脏大小没有明显差异,而SHP-2D61G/+小鼠脾脏明显增大;在SHP-2D61G/+小鼠中敲除Gab2后,SHP-2D61G/+/Gab2-/-小鼠脾指数明显低于SHP-2D61G/+小鼠(P<0.05),见图1,提示Gab2敲除能减轻SHP-2突变导致的小鼠脾脏增大。
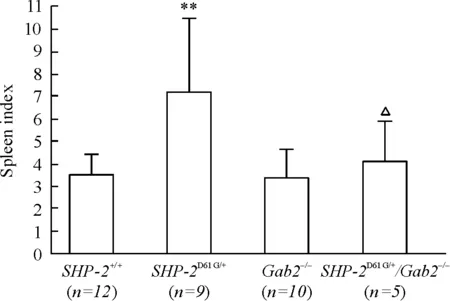
Figure 1. The spleen index ofSHP-2D61G/+/Gab2-/-mice obviously decreased, compared withSHP-2D61G/+mice. Mean±SD.**P<0.01vsSHP-2+/+;△P<0.05vsGab2-/-.
图1Gab2敲除后,SHP-2D61G/+/Gab2-/-小鼠脾指数比SHP-2D61G/+小鼠明显下降
2敲除Gab2降低SHP-2突变导致的小鼠异常增加的外周血白细胞
4组小鼠外周血白细胞计数结果显示,与SHP-2+/+小鼠相比,Gab2敲除小鼠外周血白细胞数没有明显变化,SHP-2D61G/+小鼠外周血白细胞数明显增加,而SHP-2D61G/+/Gab2-/-小鼠白细胞数明显低于SHP-2D61G/+小鼠(P<0.05),见图2,提示Gab2敲除能降低SHP-2突变导致的小鼠外周血白细胞异常增加。
3敲除Gab2降低SHP-2突变小鼠骨髓细胞中髓系比例
Gab2敲除后,SHP-2激活突变导致外周血白细胞以及骨髓细胞中髓系异常增加的现象均得到改善。图3显示SHP-2D61G/+/Gab2-/-小鼠骨髓Mac-1和Gr-1阳性细胞比例明显低于SHP-2D61G/+组小鼠(外周血结果相同,未列出)。
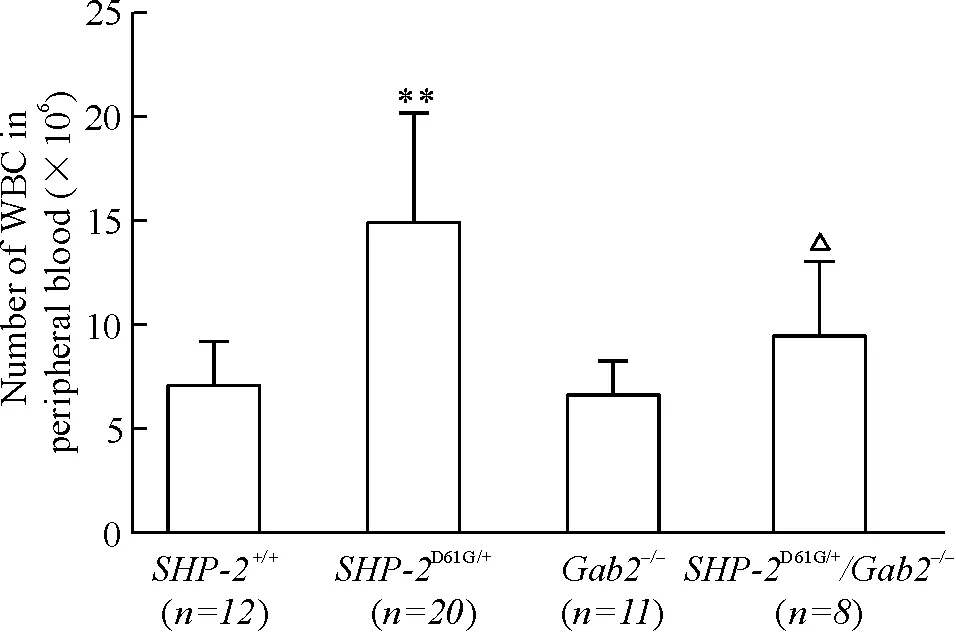
Figure 2.Gab2 knockout reduced the number of peripheral white blood cells that increased by theSHP-2D61G/+mutation. Mean±SD.**P<0.01vsSHP-2+/+;△P<0.05vsGab2-/-.
图2敲除Gab2降低SHP-2D61G/+突变导致的外周血白细胞异常增高
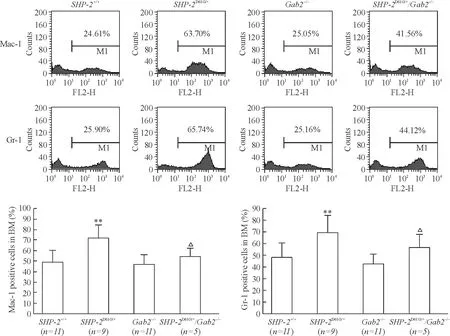
Figure 3. The proportion of Mac-1 and Gr-1 positive cells obviously decreased in the bone marrow (BM) cells ofSHP-2D61G/+/Gab2-/-mice. Mean±SD.**P<0.01vsSHP-2+/+;△P<0.05vsGab2-/-.
图3SHP-2D61G/+/Gab2-/-小鼠骨髓细胞Mac-1和Gr-1阳性比例明显低于SHP-2D61G/+小鼠
4SHP-2D61G/+/Gab2-/-小鼠骨髓细胞对IL-3的反应性降低
在甲基纤维素半固体培养系统中,用不同浓度IL-3 (0、0.2、2和10 μg/L)培养骨髓细胞,可以刺激骨髓造血祖细胞或干细胞向髓系分化增殖并形成集落。如图4所示,经IL-3刺激后,Gab2敲除小鼠骨髓细胞对IL-3的反应性降低,集落形成小而少;SHP-2D61G/+/Gab-2-/-小鼠来源的骨髓细胞集落形成能力明显低于SHP-2D61G/+单一突变的小鼠骨髓细胞,在IL-3浓度为2 μg/L时尤为显著。
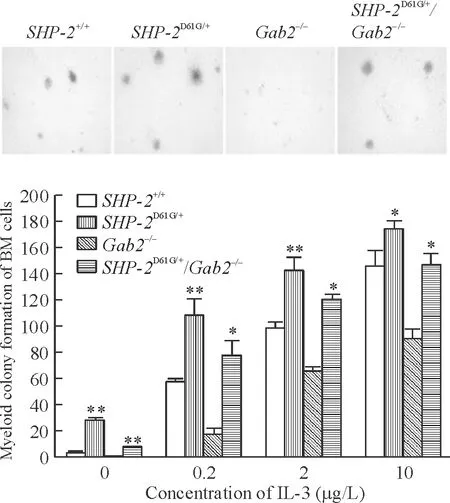
Figure 4.Gab2 knockout reduced the hypersensitivity of myeloid progenitor cells inSHP-2D61G/+mutation mice induced by IL-3. Mean±SD.*P<0.05,**P<0.01vsSHP-2+/+.
图4与SHP-2D61G/+小鼠相比,SHP-2D61G/+/Gab2-/-小鼠骨髓细胞对IL-3的反应性降低
5Gab2基因敲除后,SHP-2D61G/+蛋白无法与Gab2蛋白结合从而阻断下游信号途径活化
SHP-2在细胞信号调控过程中可以和Gab2结合,对MAPK及PI3K信号途径活化起重要作用。 实验发现,SHP-2激活突变的细胞在IL-3刺激下,SHP-2与Gab2结合增加,磷酸化ERK和Akt水平增高;Gab2敲除阻断了SHP-2D61G/+与Gab2结合,与SHP-2D61G/+小鼠相比,SHP-2D61G/+/Gab2-/-小鼠的肥大细胞在IL-3刺激下,ERK和Akt磷酸化水平降低,见图5、6。
讨 论
我们前期研究发现SHP-2D61G/+模型小鼠出现明显的髓系增殖现象,主要表现在:脾脏明显增大,外周血白细胞增多,且外周血和骨髓细胞中髓系细胞比例明显升高,其机制可能与SHP-2激活突变后导致的与细胞增殖密切相关的MAPK及PI3K信号途径异常活化有关[17-18]。 但SHP-2是酪氨酸磷酸酶,其激活突变引起的磷酸酶活性升高理论上应该导致细胞内酪氨酸磷酸化蛋白水平降低,多种细胞信号活性可能会受到抑制。为何在SHP-2D61G/+细胞内出现下游信号的增强呢,这可能与SHP-2还具有接头蛋白功能有关。 激活突变的SHP-2可以结合更多的Gab2,进而活化Ras-ERK和PI3K-Akt信号通路[6,17-18]。我们设想,除了抑制激活突变SHP-2的功能外,抑制Gab2的功能有可能减轻SHP-2激活突变导致的髓系异常增殖。为此,我们建立了SHP-2+/+、Gab2-/-、SHP-2D61G/+和SHP-2D61G/+/Gab2-/-4种基因型小鼠,观察模型小鼠髓系增殖情况,果然发现SHP-2D61G/+/Gab2-/-小鼠无论是脾脏增大、外周血白细胞增多及髓系细胞比例都较SHP-2D61G/+小鼠明显改善,Gab2缺失使SHP-2激活突变的骨髓细胞对IL-3反应性也明显降低。IP结果证实,Gab2缺失阻断了骨髓来源的肥大细胞(经IL-3刺激)内SHP-2与Gab2的结合,且ERK和Akt 的活化也明显减弱。这些结果表明,当SHP-2与Gab2的结合被阻断后,SHP-2激活突变导致的小鼠髓系异常增殖现象明显减轻,并且ERK和Akt的磷酸化水平也显著降低。这提示在SHP-2激活突变导致的小鼠髓系异常增殖过程中Gab2起到关键的接头蛋白作用,这种作用可能还会参与SHP-2激活突变相关肿瘤的发生与发展。本研究结果揭示了SHP-2激活突变导致髓系增殖等疾病的分子机制,也为SHP-2激活突变相关白血病的治疗提供一个潜在的作用靶点。

Figure 5. The binding capacity of SHP-2D61G/+with Gab2.
图5SHP-2D61G/+蛋白能与Gab2蛋白结合
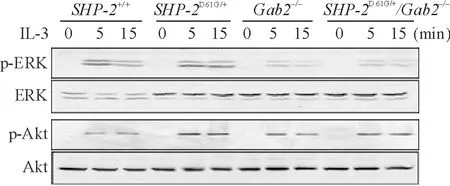
Figure 6.Gab2 knockout reduced the phosphorylation levels of ERK and Akt inSHP-2D61G/+mast cells.
图6Gab2敲除后,SHP-2突变导致的ERK和Akt活化减弱
[1] Hamidah NH, Farisah NR, Azlinda AB, et al. A study of JAK2 (V617F) gene mutation in patients with chronic myeloproliferative disorders[J]. Clin Ter, 2012, 163(2):109-113.
[2] Abraham S, Salama M, Hancock J, et al. Congenital and childhood myeloproliferative disorders with eosinophilia responsive to imatinib[J]. Pediatr Blood Cancer, 2012, 59(5):928-929.
[3] De T, Prabhakar P, Nagaraja D, et al. Janus kinase (JAK) 2 V617F mutation in Asian Indians with cerebral venous thrombosis and without overt myeloproliferative disorders[J]. J Neurol Sci, 2012, 323(1-2):178-182.
[4] Nabinger SC, Chan RJ. Shp2 function in hematopoietic stem cell biology and leukemogenesis[J]. Curr Opin Hematol, 2012, 19(4):273-279.
[5] Tartaglia M, Zampino G, Gelb BD. Noonan syndrome:clinical aspects and molecular pathogenesis[J]. Mol Syndromol, 2010, 1(1):2-26
[6] Xu D, Wang S, Yu WM, et al. A germline gain-of-function mutation inPtpn11 (SHP-2) phosphatase induces myeloproliferative disease by aberrant activation of hematopoietic stem cells[J]. Blood, 2010, 116(18):3611-3621.
[7] Xu D, Liu X, Yu WM, et al. Non-lineage/stage-restricted effects of a gain-of-function mutation in tyrosine phosphatasePtpn11 (Shp2) on malignant transformation of hematopoietic cells[J]. J Exp Med, 2011, 208(10):1977-1988.
[8] Qu CK. The SHP-2 tyrosine phosphatase: signaling me-chanisms and biological function[J]. Cell Res, 2000, 10(4): 279-288.
[9] Qu CK, Nguyen S, Chen J, et al. Requirement of SHP-2 tyrosine phosphatase in lymphoid and hematopoietic cell development[J]. Blood, 2001, 97(4): 911-914.
[10] Mohi MG, Neel BG. The role of Shp2 (PTPN11) in cancer[J]. Curr Opin Genet Dev, 2007, 17(1): 23-30.
[11] Wang S, Yu WM, Zhang W, et al. Noonan syndrome/leukemia-associated gain-of-function mutations in SHP-2 phosphatase (PTPN11) enhance cell migration and angiogenesis[J]. J Biol Chem, 2009, 284(2): 913-920.
[12] Grossmann KS. The tyrosine phosphatase Shp2 in development and cancer[J]. Adv Cancer Res, 2010, 106: 53-89.
[13] Martinelli S, Nardozza AP, Delle Vigne S, et al. Counteracting effects operating on Src homology 2 domain-containing protein tyrosine phosphatase 2 (SHP2) function drive selection of the recurrent Y62D and Y63C substitutions in Noonan syndrome[J]. J Biol Chem, 2012, 287(32):27066-27077.
[14] Liu X, Qu CK. Protein tyrosine phosphatase SHP-2 (PTPN11) in hematopoiesis and leukemogenesis[J]. J Signal Transduct, 2011,2011:195239.
[15] Araki T, Mohi MG, Ismat FA, et al. Mouse model of Noonan syndrome reveals cell type- and gene dosage-dependent effects ofPtpn11 mutation[J]. Nat Med, 2004, 10(8):849-857.
[16] Yu WM, Daino H, Chen J, et al. Effects of a leukemia-associated gain-of-function mutation of SHP-2 phosphatase on interleukin-3 signaling[J]. J Biol Chem, 2006, 281(9):5426-5434.
[17] 张 薇,杨金莲,胡中倩,等.SHP-2酪氨酸磷酸酶激活突变导致小鼠髓系异常增殖[J].中国病理生理杂志, 2011,27(4):682-687.
[18] 霍寅萍,储著朗,余科科,等.SHP-2酪氨酸磷酸酶激活突变的肥大细胞对IL3呈高增殖敏感性[J].安徽医科大学学报, 2010, 45(5):593-596.
[19] Nishida K, Yamasaki S, Hasegawa A, et al. Gab2, via PI-3K, regulates ARF1 in FcεRI-mediated granule translocation and mast cell degranulation[J]. J Immunol, 2011, 187(2):932-941.
[20] Vaughan TY, Verma S, Bunting KD. Grb2-associated binding (Gab) proteins in hematopoietic and immune cell biology[J]. Am J Blood Res, 2011, 1(2):130-134.
[21] Futami M, Zhu QS, Whichard ZL, et al. G-CSF receptor activation of the Src kinase Lyn is mediated by Gab2 recruitment of the Shp2 phosphatase[J]. Blood, 2011, 118(4):1077-1086.
RoleofGab2inmousemyeloidabnormalproliferationinducedbySHP-2D61G/+mutation
CHEN Ji1, WANG Xin-yi1, CHEN Zhuo1, LI Fei-fei1, ZHENG Hong1, QU Cheng-kui1, 2, WANG Si-ying1
(1DepartmentofPathophysiology,AnhuiMedicalUniversity,Hefei230032,China;2CaseWesternReserveUniversity,Cleveland,Ohio44106,USA.E-mail:sywang@ahmu.edu.cn)
AIM: To investigate whether Gab2, the key adapter protein in the SHP-2 signaling pathway, is involved in mouse myeloid abnormal proliferation induced bySHP-2D61G/+mutation.METHODSFour kinds of mouse model genotyped asSHP-2+/+,Gab2-/-,SHP-2D61G/+andSHP-2D61G/+/Gab2-/-were generated from crossbreeding ofGab2-/-mice andSHP-2D61G/+mice. The mouse spleen size was analyzed. The number of peripheral blood leukocytes was counted by cell counting and the percentage of Mac-1 or Gr-1 positive myeloid cells in the bone marrow was detected by flow cytometry. The proliferation ability of bone marrow hematopoietic stem/progenitor cells in response to cytokines was assayed by colony formation. The expression of p-ERK and p-Akt and the binding capacity of SHP-2 with Gab2 in the bone marrow-derived mast cells stimulated with IL-3 were detected by Western blotting and immunoprecipitation.RESULTSThe phenotype of myeloproliferative disorder, such as enlarged spleen size, increased leukocyte number and high percentage of myeloid cells, inSHP-2D61G/+mutant mice was found, and was dramatically improved inSHP-2D61G/+/Gab2-/-double mutation mice. Furthermore, compared withSHP-2D61G/+mutation mice, significantly decreased colony formation ability of the bone marrow cells with IL-3 stimulation was observed inSHP-2D61G/+/Gab2-/-double mutation mice. A reduced phosphorylation level of ERK/Akt, and SHP-2 without binding of Gab2 were found inSHP-2D61G/+/Gab2-/-bone marrow-derived mast cells with IL-3 stimulation.CONCLUSIONGab2 knockout significantly reduces mouse myeloid abnormal proliferation induced bySHP-2D61G/+mutation. The molecular mechanism may be associated with reduced binding ofSHP-2D61G/+underGab2 knockout, and further weakened the activation of downstream signaling pathways of ERK and Akt.
SHP-2 tyrosine phosphatase; Genetic gain-of-function mutation; Gab2; Myeloproliferative disorders
R363
A
10.3969/j.issn.1000- 4718.2013.06.008
1000- 4718(2013)06- 1003- 06
2012- 12- 28
2013- 04- 17
国家自然科学基金资助项目(No. 30873046; No.30973424; No.81272258); 教育部博士点基金资助项目(No. 200803660005);安徽省出国留学回国人员科技项目(N0. 2009-2011)
△通讯作者 Tel: 0551-65137833; E-mail: sywang@ahmu.edu.cn
猜你喜欢
中国典型病例大全(2022年9期)2022-04-19
中国药学药品知识仓库(2022年1期)2022-03-23
世界复合医学(2021年9期)2021-11-26
中国毕业后医学教育(2020年4期)2020-12-06
中华临床免疫和变态反应杂志(2018年6期)2019-01-17
同济大学学报(医学版)(2018年4期)2018-09-10
课程教育研究(2017年31期)2017-09-15
中国骨质疏松杂志(2017年7期)2017-08-07
中国科技纵横(2017年12期)2017-07-25
中国医药导报(2017年4期)2017-04-06
