
Mn3O4多面体纳米晶体的制备及其电化学性能
2013-10-17 03:03杨陆峰郑明涛胡超凡崔江虎刘应亮
无机化学学报 2013年2期
杨陆峰 高 闯 郑明涛 胡超凡 崔江虎 刘应亮*,
(1暨南大学化学系纳米化学研究所,广州 510632)
(2华南农业大学理学院,广州 510642)
The development of manganese oxides nanocrystals has been intensively pursued due to their useful applications in the areas of catalysis,energy storage,chemicalsensing devices,magneticdata storage and ferro-uids[1-4].Among them,hausmannite Mn3O4presents particular interest because of its application as an effective catalyst for the decomposition of waste gas and waste solution[5-6].Recent studies have shown that nanostructural Mn3O4possesses interesting electrochemical properties.For instance,Zhang et al.[7]prepared Mn3O4polyhedron nanocrystal via thermolysis of a hydrogen-bonded polymer,which exhibitsa betterelectrochemical capacitance performance than spinel Mn3O4layered nanostructure[8].The performance of the metal oxides is dependent on the structure and morphology including crystallite size,stacking manner,orientation and aspect ratio,which are sensitive to the synthesis route of their preparation[9].
Owing to the unique shape and size-dependent properties,Mn3O4has been prepared with various methods such as solvothermal treatment of manganite(MnOOH)[10-12],solvothermal treatment of manganese acetate(Mn(CH3COO)2·4H2O)with a hydrogen-bonded polymer[7],sonochemical method to prepare sphere-like nanocrystals[13],calcination of nitrate (Mn(NO3)2),carbonate (MnCO3),manganese oxides(MnO2,Mn2O3,etc.)and oxyhydroxide(g-MnOOH)at high temperature(1 000℃)[14-16],precipitation method from manganese nitrate (Mn(NO3)2)at moderate temperature[17],sol-gel process with a post-treatment at higher temperature[18-19],chemical bath deposition to prepare thin lms[20],electro spinning technique[21],gas condensation[22].
However,synthetic methods that take advantage of costly organic surfactants and templates often require subsequent purification procedures which markedly increase manufacture costs[23-24].The preparation of Mn3O4by using the precursor manganese oxides with the sophisticated instrument could not be afforded by the average laboratories in the practical application.In the meanwhile,owing to the complex inuence of pH value,temperature,ion concentration,and so forth,limited breakthrough has been achieved for the morphology and size-controlled synthesis ofnanostructured Mn3O4through the solution based approach without any additives.Therefore,it is of great significance to develop an environmentally friendly,low-cost and template-free synthetic method for the synthesis of Mn3O4nanostructures.
In our previous work,we have reported a simple method for the controlled synthesis of uniform shaped carbon hollow structures by an ethanol-assisted thermolysis of zinc acetate[25],which uses the generated zinc oxide nanostructures as in-situ templates.Herein,we report a one-step synthesis of Mn3O4polyhedral nanocrystals through a facile solution-based thermolysis route in the manganese acetate-alcohol system without any additives.In addition,the formation mechanism of the products has also been proposed.The electrochemical properties of the optimized products of Mn3O4nanocrystals were examined by cyclic voltam metry(CV)measurements.
1 Experimental
1.1 Synthesis of Mn3O4polyhedral nanocrystals
All chemicals used were of analytical grade and purchased from the commercial market without further purification.The synthesis of Mn3O4nanostructures was carried out via a solvothermal method.In a typical procedure,2.5 mmol Mn(CH3COO)2·4H2O(Mn(Ac)2)were dissolved in 30 mL of absolute ethanol.Then the mixture was stirred to give a clear solution and transferred into a 45 mL Teflon-lined stainless steel autoclave.The autoclave was maintained at 200 ℃ for 1~24 h,with a heating rate of 10℃·min-1in an electronic furnace,and then cooled naturally to room temperature.The resulting precipitate was centrifuged and thoroughly washed with deionized water and ethanol several times before drying in air at 60℃ for 24 h.
1.2 Characterization
Crystallographic phases of the products were examinedbyXRD usingaMSALXD2X-ray diffractometer with Cu Kα radiation(36 kV,20 mA,λ=0.154 18 nm).FTIR spectra were measured by an Equinox 55 (Bruker)spectrometer with the KBr pellet technique from 400 to 4 000 cm-1.Raman spectra of sampleswere measured using a Renishaw Via microspectrometer using an excitation wavelength of 514 nm generated by an Ar+laser.A 100× objective was used to focus the laser beam and to collect the Raman signal.Chemical state analysis was carried out by X-ray photoelectron spectroscopy (XPS)using a Shimadzu AXIS Ultra X-ray photoelectron spectrometer.All XPS spectra were corrected using the C1s line at 284.6 eV.Casa XPS Version 2.3.13 software and Origin Pro 8.0 were used to analyze the experimental data and graphs.The morphologies of the samples were characterized with scanning electron microscopy (SEM,PhilipsXL-30s),transmission electron microscopy (TEM,Philips Tecnai-10)and high-resolution TEM (HR-TEM,GEOL-2010).The typical components of the liquid fractions after the reaction were detected by the gas chromatography and mass spectrometry(GC-MS)technique.The conditions for GC-MS are given in the supporting information.
1.3 ElectrochemicalmeasurementofMn3O4 electrode
The electrochemical electrode was tested on a CHI 660B electrochemical workstation in a three-electrode system.The working electrode was fabricated by pressing the mixture of Mn3O4,carbon black and 5%-PTFE(Polytetrafluoroethylene)(75∶15∶10,W/W/W)into foam nickel electrode.A Pt slice was used as auxiliary electrode and a Ag/AgCl as reference electrode.In these studies,all measurements were performed in a 0.5 mol·L-1Na2SO4aqueous electrolyte solution and all electrochemical experiments were carried out at room temperature.
2 Results and discussion
2.1 Characterization of materials
The crystalline structure and phase purity of the product were investigated by XRD as shown in Fig.1a.Mn3O4are synthesized with 2.5 mmol Mn(Ac)2under the present condition,which can be easily indexed to pure tetragonal phase of γ-Mn3O4(PDF No.89-4837)with lattice constants a of 0.576 3 nm and c of 0.945 6 nm.Comparatively,with the coexistence of γ-Mn3O4,the sample prepared with 7.5 mmol Mn(Ac)2exhibits strong diffraction peak of MnO (PDF No.89-4835)(Fig.1b),indicating that the reaction dosage of Mn(Ac)2has vital influence on the crystals structure of the product.
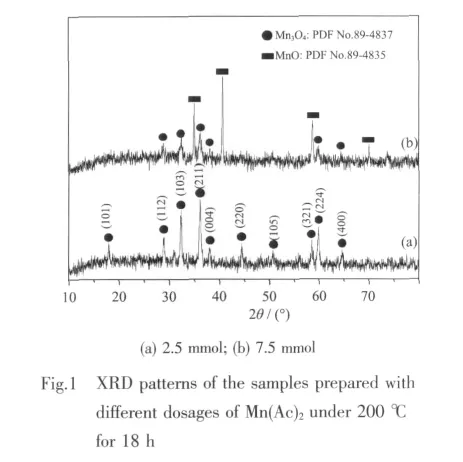
The FTIR spectrum in Fig.2 provides more convincing evidence of pure Mn3O4fabricated with 2.5 mmol Mn(Ac)2,which displays three characteristic peaks at 638,532 and 416 cm-1[26-27].Besides,strong absorptions at 3 442 cm-1and weak absorptions around 2 800~3 000 cm-1respectively reveal the stretching vibrations of O-H and C-H.The absorptions at 1 635,1 384 and 1 112 cm-1correspond to the vibrations of CO,C-H groups.Therefore,the FTIR spectrum suggests that the surface of the nanoparticles is coated by a layer of ethanol molecules.
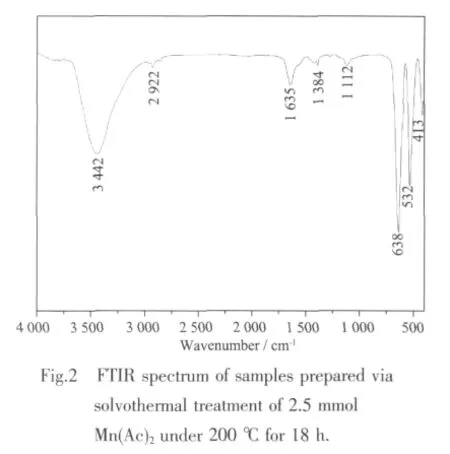
Considering the C-OH and C-H vibration,and Ac-/C2H5OH used in hydrothermal conditions,the Raman spectra have been provided to investigate the surface microstructure of the pure Mn3O4nanocrystals,as shown in Fig.3.From the Raman spectrum,the Raman peaks at 652.3 cm-1corresponding to crystalline hausmannite structure are clearly found,which are in good agreement with the microstructure information of as-prepared Mn3O4[28-29].In addition,there are no diffraction peaks around~1 360 cm-1and 1 580 cm-1,suggesting the absence of carbon layer on the Mn3O4prepared under the hydrothermal conditions at 200℃.
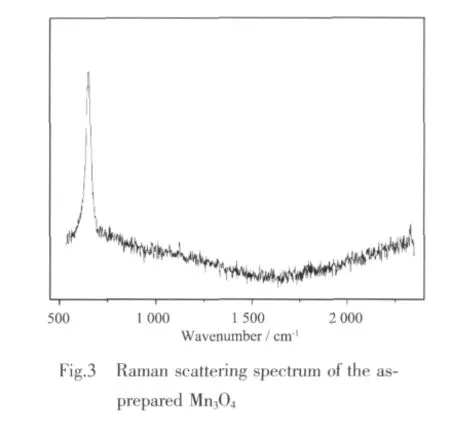
Chemical state information for the as-prepared Mn3O4was studied using XPS.As shown in Fig.4a,the survey spectrum shows no significant presence of impurities,except for the contaminant carbon.Energy levels of Mn3s,O1s,Mn2p and Mn3p are obtained in the exact energy locations as reported earlier[30-31].In Fig.4b,the binding energy value of Mn2p3/2is 641.6 eV,and the spin orbit splitting between the Mn2p3/2and Mn2p1/2level is 11.7 eV,which perfectly matches the previously reported values for hausmannite[32].The oxidation state of the manganese atom is further analyzed by deconvoluted for relative intensities of the component peaks of 3p3/2XPS peaks.The binding energy of the Mn2p3/2peak components (640.9 and 642.6 eV)is in good agreement with the literature report,respectively,for the occurrence of Mn2+and Mn4+[31,33]for the formation of 2MnO-MnO2.The atomic concentration of the total oxygen and the manganese from the results of XPS is 64.5% and 35.5%,respectively.The atomic ratio of O to Mn in the Mn3O4is 1.82,which is greater than the theoretical value of 1.33.The excess O may come from the ethanol molecules coated on the surface ofMn3O4as demonstrated by the FTIR spectrum.
In order to study the inuence of the reaction time on the morphology ofthe products,series of experiments were carried out.SEM images in Fig.5a shows a low-magnication of the sample obtained after solvothermal reaction at 200℃for 2 h,which exhibit large-scale formation of uniform nanoparticles with diameters about 10 ~20 nm.The nanometer-sized particles are observed with diameter of 40~60 nm aggregative attached together in Fig.5b.With increasing the reaction time to 24 h,many polyhedral Mn3O4nanocrystals with mean diameter of 250 nm are presented in Fig.5c.Therefore,the SEM images of the samples reveal that the reaction time plays vital role in the shape evolution of Mn3O4.When the reaction time is extended,the size of Mn3O4sample grows bigger and exhibits distinct polyhedral nanocrystals.

For the reason that the poor resolution of SEM images can not throw light on the formation process of Mn3O4,the morphology and microstructures of the asprepared Mn3O4were further investigated with TEM and HRTEM.As shown in Fig.6a,a mass of uniform tiny nanoparticles with average diameter of 8 nm are observed after reaction of 1 h.When the reaction time is increased to 2 h,it can be clearly observed that the initial nanocrystals grow into uniform hexagonal flakelike morphology with widths of 10~15 nm as shown in Fig.6b.When the reaction time is further increased to 12 h,many polyhedral nanocrystals with rough surface are observed,which project to regular tetragonal shape with edge lengths of 40~60 nm(Fig.6c).
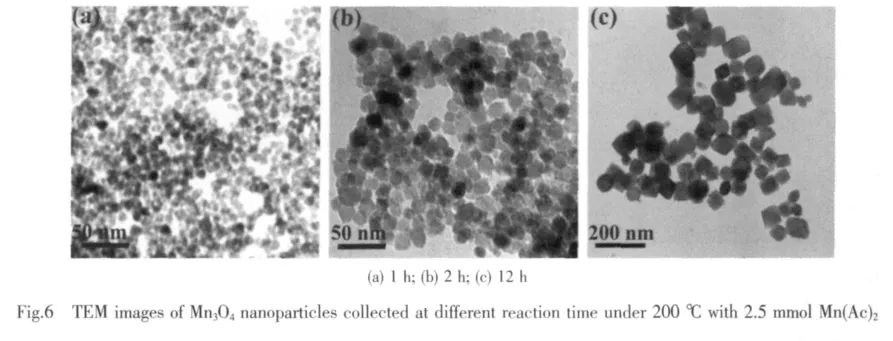
The uniform polyhedral nanocrystals with smooth surface are produced when the reaction time is extended to 18 h (Fig.7a).In the same time,the corresponding XRD pattern exhibits sharp and strong diffraction peaks of γ-Mn3O4(Fig.1a).More details of the structure are investigated by HRTEM images.Homogeneous octahedral like nanocrystals with edge lengths about 120 nm are clearly seen in Fig.7b.As shown in Fig.7c,the corresponding lattice fringes exhibit distinct sets of lattice spacing of about 0.306 nm,consistent with(112)crystal planes of a tetragonal structure.Therefore,the Mn3O4nanostructure can be well controlled by tuning the reaction time at 200℃with 2.5 mmol Mn(Ac)2.The time-dependent experiments reveal that the formation of Mn3O4nanocrystals experience an Ostwald ripening dominated process,which is well consistent with the SEM investigation.
2.2 Formation mechanism of polyhedral nanocrystals

X-ray crystallography analysis shows that Mn3O4is obtained with 2.5 mmol Mn(Ac)2through the solvothermal process.Interestingly,the synthesized product exhibits the crystalline phase of MnO when increasing the dose of Mn(Ac)2to 7.5 mmol at 200 ℃ (Fig.1b).Therefore,it can be deduced that there is an oxidizing process of MnⅡto MnⅢ with O2both in the autoclave and the ethanol solution.And the limited amount of O2in the reaction system is not enough to have 7.5 mmol MnⅡall oxidized into MnⅢ.Meanwhile,an intense odor of ester is noticed from the solution after the reaction.To shed light on the formation process of Mn3O4,the reaction by-product has been investigated via GC/MS technique.GC/MS analysis conducted on the extract of the post-reaction solution clearly reveals the existence of ethyl acetate (Fig.S1,S2,Supporting Information),indicating the formation of butyl acetate during the synthetic procedure,which is similar to the previous report[34].Thus,On the basis of the experimental results,a formation mechanism of Mn3O4polyhedral nanocrystals is proposed.We believe that the reaction process of the manganese acetate-alcohol solution may be as follows:
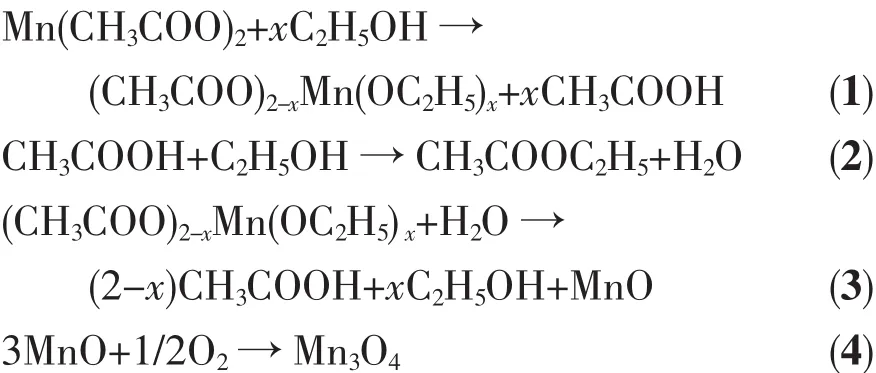
In the current solvothermal synthesis at 200℃,the reaction between manganese acetate and alcohol firstly results in the coordination of C2H5OH to manganese centers,to form unstable alcohol acetate complexes(CH3COO)2-xMn(OC2H5)xby ligand exchange/substitution,concomitant with the release of CH3COOH(eq.1).The produced acetic acid could then react with the solvent alcohol to form water by a slow esterification reaction(eq.2).Subsequently,(CH3COO)2-xMn(OC2H5)xwould hydrolyze and generate MnO under the selfgenerated pressure (eq.3).Finally,Mn3O4nanostructures are achieved after the oxidation reaction of the active MnO with the O2both in the reaction container and ethanol solution (eq.4).In this current situation,many newly formed Mn3O4colloids aggregate together andform nuclei.Meanwhile,thenascentMn3O4nanocrystals with high surface energies might be temporarily stabilized by ethanol solution.Then,the dissolution recrystallization and self-assembly process dominate.Subsequently,an Ostwald ripening process dominates.The proposed pathway is supported by our time-dependent experiments,as shown in Fig.6a~c.
2.3 Electrochemical properties of Mn3O4 polyhedral nanocrystals
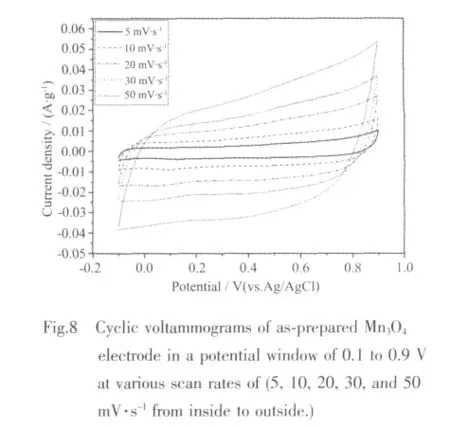
The electrochemical performance of Mn3O4polyhedral nanocrystals synthesized at 200℃for 18 h were evaluated as a supercapacitor electrode in view of their intrinsic properties and unique structural features.Fig.8 shows the cyclic voltammetry (CV)analysis at various scan rates in 0.5 mol·L-1Na2SO4electrolyte with a potential range of 0.1 to 0.9 V.(vs.Ag/AgCl).The CV curves at slow scan rate present an ideal capacitive behavior with ne rectangular shape.The deviation from rectangularity of the CV becomes distinct with the increase of scan rate.No obvious redox peaks are present in the CV curves,revealing that the measured electrode is charged and discharged at a pseudo-constant rate over the complete voltammetric cycle[35].
The MnO2based electrode[36]reveal maybe the charge storage mechanism in Mn3O4electrode.At slower scan rate,almost all available pores both on the surface and inside of Mn3O4electrode can be filled with Na+from electrolyte,resulting in a better effective utilization of Mn3O4for redox reaction and a better capacitance.However,the effective interaction between the ions and the electrode is greatly decreased when increasing the scan rate.Thus,the effective redox reaction of Mn3O4is confined only to the outer surface of Mn3O4electrode,resulting in decreased capacitance.The specic capacitance of the electrode at different current densities can be calculated from the following equation[37]
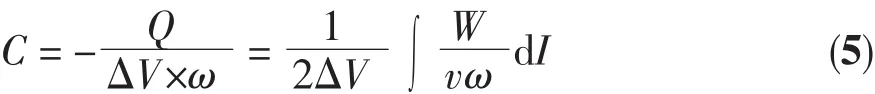
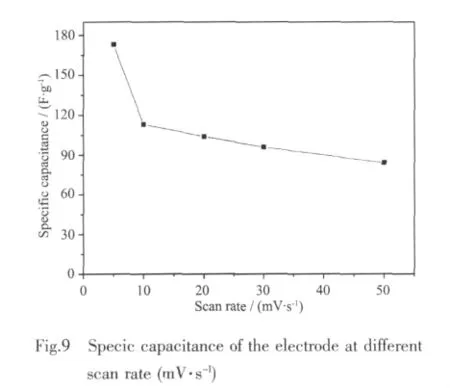
where C is the specific capacitance of the electrode based on the mass of active materials(F·g-1),Q is the sum of anodic and cathodic voltammetric charges on positive and negative sweeps(C),I is the sample current(A),W is the weight of active materials(g),and V is the total potential deviation of the voltage window(V).v is the scanning rate(V·s-1)and ω is the mass of active electrode materials (g).According to formula(1),specic capacitance ranging from 173 to 84 F·g-1can be delivered at the various scan rates of 2~50 mV·s-1,as shown in Fig.8,which is a little lower than that of Mn3O4nano-octahedrons[38].Thus,the superior crystal structure should be provided for a better functional properties of Mn3O4。
3 Conclusions
In summary,Mn3O4nanocrystals with wellcontrolled polyhedral shape have been synthesized via Ethanol-assisted thermolysis of 2.5 mol manganese acetate at 200℃ for 18 h.On the basis of the results,a formation mechanism of Mn3O4polyhedral nanocrystals is proposed.It is valuable to study the shape evolution of γ-Mn3O4in the present reaction system for the understanding of the formation process of polyhedron structures.Cyclic voltammetry measurement shows that the as prepared Mn3O4electrode exhibits a good pseudo capacitance behavior with a discharge specific capacitance of 173 F·g-1at a sweep rate of 5 mV·s-1.These results suggest that Mn3O4polyhedral nanocrystals materials may have potential applications in electrochemical capacitor.
[1]Zarur A J,Ying J Y.Nature,2000,403:65-67
[2]Majetich S A,Jin Y.Science,1999,284:470-473
[3]Nayral C,Viala E,Fau P,et al.Chem.Eur.J.,2000,6:4802-4090
[4]Raj K,Moskowitz R.J.Magn.Magn.Mater.,1990,85:233-245
[5]Stobhe E R,Boer B A,Geus J W.Catal.Today,1999,47:161-167
[6]Yamashita T,Vannice A.J.Catal.,1996,163:158-168
[7]Zhang F,Zhang X G,Hao L.J.Mater.Chem.Phys.,2011,126:853-858
[8]Dai Y,Wang K,Xie J Y.Appl.Phys.Lett.,2007,90:102-104
[9]Jiang J,Li L.Mater.Lett.,2007,61:4894-4896
[10]Zhang W,Yang Z,Liu Y,et al.J.Cryst.Growth,2004,263:394-399
[11]Demazeau G.J.Mater.Chem.,1999,9:15-18
[12]Walton R I.Chem.Soc.Rev.,2002,31:230-238
[13]Askarinejada A,Bagherzadehb M,Morsali A.J.Appl.Surf.Sci.,2010,256:6678-6682
[14]Shomate C H.J.Am.Chem.Soc.,1943,65:786-789
[15]Southard J C,Moore G E.J.Am.Chem.Soc.,1942,64:1769-1770
[16]Ursu I,Alexandrescu R,Mihailescu I N.J.Phys.B,1986,19:825-830
[17]Rabiei S,Miser D E,Lipscomb J A,et al.J.Mater.Sci.,2005,40:4995-4998
[18]Ching S,Roark J L,Duan N.Chem.Mater.,1997,9:750-754
[19]Al Sagheer F A,Hasan M A,Pasupulety L.J.Mater.Sci.Lett.,1999,18:209-211
[20]Xu H Y,Xu S L,Wang H,et al.J.Electrochem.Soc.,2005,12:803-807
[21]Shao C,Guan H,Liu Y,et al.J.Solid State Chem.,2004,177:2628-2631
[22]Dimesso L,Heider L,Hahn H.Solid State Ionics,1999,123:39-46
[23]Manna L,Milliron D J,Meisel A,et al.Nat.Mater.,2003,2:382-385
[24]Tian Z R,Voigt J A,Liu J,et al.Nat.Mater.,2003,2:821-826
[25]Zheng M T,Liu Y L,Zhao S,et al.Inorg.Chem.,2010,49:8674-8683
[26]Yang B J,Hu H M,Li C,et al.Chem.Lett.,2004,33:456-458
[27]Ocana M.Colloid.Polym.Sci.,2000,278:443-449
[28]Wang W Z,Ao L.Cryst.Growth Des.,2008,8:358-362
[29]Zuo J,Xu C,Qian Y T,et al.Nanostruct.Mater.,1998,10:1331-1335
[30]Zhao L Z,Young V.J.Electron Spectrosc.Relat.Phenom.,1984,34:45-54
[31]Ezhil Raj A M,Victoria S G,Jothy V B,et al.J.Appl.Surf.Sci.,2010,256:2920-2926
[32]Foord J S,Jackman R B,Allen G C.Philos.Mag.A,1984,49:657-663
[33]Castro V D,Polzonetti G.J.Electron Spectrosc.,1989,48:117-123
[34]Ye J F,Liu W,Cai J G.J.Am.Chem.Soc.,2011,133:933-940
[35]Xu M W,Kong L B,Zhou W J,et al.J.Phys.Chem.C,2007,111:19141-19147
[36]Devaraj S,Munichandraiah N.J.Phys.Chem.C,2008,112:4406-4417
[37]YI Guan-Gui(易 观 贵 ),XIAO Yong(肖 勇 ),HE Wen-Qi(贺文启),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2011,27(1):162-166
[38]Jiang H,Zhao T,Yan C,et al.Nanoscale,2010,2:2195-2198
猜你喜欢
声屏世界(2022年21期)2023-01-07
华南农业大学学报(2022年6期)2022-10-31
数学大王·低年级(2022年3期)2022-03-17
课外生活·趣知识(2021年8期)2021-08-24
中学课程辅导·高考版(2020年12期)2020-12-23
科技进步与对策(2020年22期)2020-12-08
科技进步与对策(2020年10期)2020-06-30
———理学院
西安航空学院学报(2018年1期)2018-02-05
杭州师范大学学报(自然科学版)(2017年3期)2017-06-15
金色年华(2016年11期)2016-02-28
