
儿童Castleman 病9 例临床分析
2013-09-10 02:59宋红梅张伟宏师晓华
中国循证儿科杂志 2013年4期
李 卓 肖 娟 宋红梅 沈 菁 魏 珉 张伟宏 师晓华
Castleman 病( CD) 又称巨大淋巴结增生症或血管滤泡性淋巴样增生症,是一种介于良恶性之间的淋巴增殖性疾病,一般认为是一种原因不明的慢性非特异性炎症反应。临床上主要表现为单个淋巴结肿大或者全身多发淋巴结肿大,据此可分为单中心型( UCD) 和多中心型( MCD) 。病理上可分为透明血管型、浆细胞型和混合细胞型。CD 临床较为少见,多发于成人,其中女性发病者稍多。由于临床上儿童CD 发病者罕见,且临床表现多样,缺乏特异性,漏诊和误诊率较高。目前国内有关儿童CD 的报道不多,本文对北京协和医院( 我院) 20 年来确诊的9 例CD 患儿的临床特征和预后进行回顾性分析,以期提高儿科医生对儿童CD的认识。
1 方法
1.1 对象 收集1990 年1 月至2011 年12 月在我院儿科住院,并经病理确诊为CD,且发病年龄<18 岁的儿童。
1.2 诊断标准 根据1988 年Frizzera[1]提出的标准,并结合《血液病诊断及疗效标准》第2 版[2],儿童CD 的诊断标准如下: ①UCD: a. 单一部位的淋巴结肿大,形成巨大肿块,直径3 ~7 cm,少数可达16 cm;b.组织病理学上具有特征性增生,并除外可能的原发病;c.除浆细胞型外,多无全身症状、贫血、ESR 加快和丙种球蛋白增高等; d.肿物切除后长期存活。②MCD: a. 具有特征性增生性组织病理改变;b.淋巴结肿大显著并累及多处外周淋巴结;c.伴全身症状或出现全身多系统受累表现,如长期发热、乏力和消瘦,常伴有肝脾肿大,出现贫血、高免疫球蛋白血症和低白蛋白血症等;d.排除已知可能的病因。
1.3 资料截取 从病史中截取UCD、MCD 患儿的临床表现、实验室检查、病理学检查、治疗和预后的数据,进行描述性分析。
2 结果
2.1 一般情况 9 例经手术活检和病理确诊CD 患儿进入分析,男2 例,女7 例。发病年龄在6 ~17.7 岁,中位年龄13 岁。UCD 5 例,均为女性;MCD 4 例,男2 例,女2 例。从发病到确诊时间为3 周至12 年。5 例在外院分别被误诊为结核感染( 例6,9) 、细菌感染( 例4,7,8) 、传染性单核细胞增多症( 例8) 、白塞病( 例4) 和淋巴瘤( 例8) 。
2.2 临床表现 如表1 所示,①首发症状: 淋巴结肿大5例,发热、乏力3 例,皮疹2 例,口腔溃疡1 例。②淋巴结肿大:9 例均有淋巴结的明显肿大( 淋巴结直径4 ~7 cm) ,其中浅表淋巴结肿大5 例,如颈部、腋窝和腹股沟等部位;3例为就诊后行影像学检查提示深部肿大淋巴结,如: 纵膈、腹膜后等处深部淋巴结肿大。③全身系统表现:4 例MCD患儿均伴发热,体温37.5 ~38.5℃; 并伴脾肿大,中、重度贫血;其中例8,9 有生长发育落后。④例4 为UCD 合并副肿瘤天疱疮( PNP) ,以口腔及唇黏膜糜烂、多形性皮疹起病,之后出现肺间质纤维化,腹部增强CT 示腹膜后肿大淋巴结[3]。
2.3 实验室检查指标
2.3.1 MCD ①血常规:4 例均存在中至重度贫血; WBC总数正常但中性粒细胞比例稍高,3 例PLT 在450 ~550 ×109·L-1;②骨髓象:1 例偶见Pelger-Huet 畸形,1 例为炎症性骨髓象,余2 例未见异常;③炎症指标:4 例CRP 均升高、ESR 增快;④免疫学检查:IgG 升高2/3 例;4 例血清蛋白电泳均示球蛋白比例升高。1 例抗平滑肌抗体1∶160。
2.3.2 UCD 除例6 合并PNP,余4 例血常规、骨髓象、炎症指标和免疫学指标均未见异常;例4 类风湿因子升高、抗核抗体( +) 胞浆型1∶160、抗可提取性核抗原抗体( 免疫印迹) 抗SSA 抗体( +)52KD,天疱疮抗体1∶320。
2.4 影像学检查 9 例超声或CT 等检查提示存在单一或多发淋巴结的侵犯。例2 颈部平扫CT 显示左侧颈部淋巴结增大,甲状腺的左叶及气管后受压移位( 图1A) ,增强CT显示淋巴结明显强化( 图1B) 。

图1 CD 患儿淋巴结肿大CT 所见Fig 1 CT demonstrating left enlarged cervical lymph node
2.5 病理学结果 9 例CD 患儿淋巴结均经病理切片和免疫组化染色检查,其中有5 例(71.4%) 确诊为透明血管型( UCD 4 例,MCD 1 例) ,2 例( 28.6%) 为浆细胞型( UCD和MCD 各1 例) ,无混合细胞型,2 例( UCD 和MCD 各1例) 病理切片来自于外院无法准确分型。透明血管型病理表现为淋巴结基本结构保持完整,滤泡增生明显,滤泡中心血管增生伴玻璃样变性,其间夹杂核呈空泡状的滤泡树突状细胞,生发中心周围小淋巴细胞环状排列似洋葱皮样,滤泡间及副皮质区血管增生( 图2A,B) 。浆细胞型表现为滤泡间浆细胞弥漫性增生,滤泡中玻璃样变性的血管不明显,代之以无定型的嗜酸性物质沉积(2C,D) 。免疫组化显示: CD20、CD79、CD3、CD4、CD8 等 有 不 同 程 度 表 达,CD138、VS38C 表达显示浆细胞增生为多克隆性,而非肿瘤性。EBV 阴性,异常生发中心中滤泡树突状细胞CD21 阳性。
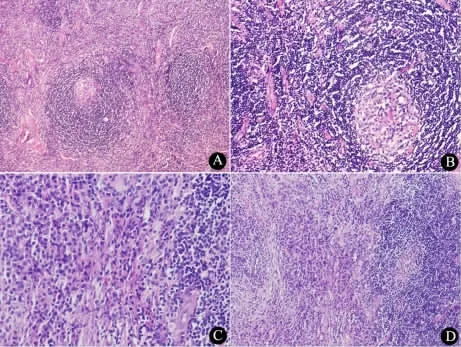
图2 CD 患儿淋巴结病理活检所见Fig 2 Pathological findings of biospy of cervical lymph node in CD patients
2.6 治疗和随诊 5 例UCD 患儿均行手术切除肿大淋巴结,其中4 例无合并症者手术后临床完全缓解,随访3 年11月至8 年10 月无复发。例4 合并PNP,经手术切除肿大淋巴结,予大剂量IVIG 冲击、激素和环磷酰胺联合治疗后,皮疹明显好转,但口腔溃疡及肺部症状仍较重,家属放弃治疗,自动出院,可能预后不佳。4 例MCD 患儿,例7 于当地医院曾接受2 个疗程不正规COP 方案化疗,疗效欠佳,于我院行CHOP 方案化疗后达临床缓解; 余3 例经手术切除肿大淋巴结及对症治疗后,其中例9 行脾切除,临床症状逐渐好转,血常规、CRP 和免疫学指标明显改善,3 例均因家属对化疗的顾虑而未行化疗,目前随访16 ~36 个月,体温正常,复查血常规、CRP 和ESR 均正常,但影像学检查仍提示有多发肿大的淋巴结。
3 讨论
CD 是一种少见的非特异性淋巴增殖性疾病,临床上表现为单发或全身多发的淋巴结肿大,该病最早报道于1954年[4],1956 年由Castleman 详细定义[5]。CD 的发病原因尚不清楚,有研究显示,IL-6 表达增加,人类疱疹毒-8( HHV-8) 、HIV 感染,免疫紊乱可能与CD 的发生有关[6]。CD 患者血清、淋巴结生发中心的B 细胞及部分滤泡树突状细胞中IL-6 表达增加,IL-6 的过度表达可诱导大鼠出现类MCD表现[7]。HHV-8 是MCD 的致病因素,HHV-8 的病毒负荷与MCD 的病情活动相关,复制中的HHV-8 可编码病毒源IL-6,促进疾病发生[8,9]。根据本文MCD 患儿均有发热、炎性指标升高等表现,2 例存在自身抗体阳性,支持免疫异常在CD 的发生中起了一定作用。
本文UCD 患儿均为女孩,病理结果均为透明血管型;MCD 患儿男女比例为1 ∶1,病理结果以浆细胞型为主。Nadia 等[10]报道成人UCD 患者男女比例约为1∶1.4,女性稍多,病理上以透明血管型为主。MCD 患者男女比例为1.8∶1,男性较多,病理上以浆细胞型为主。国内黄爽等[11]报道5 例儿童CD 中,UCD 3 例( 男1 例,女2 例) 均为透明血管型; MCD 2 例( 男女各1 例) 均为浆细胞型。Parez等[12]报道5 例UCD 患儿病理类型均为透明血管型,2 例MCD 中浆细胞型和混合细胞型各1 例; Smir 等[13]报道8例MCD 患儿,5 例为浆细胞型;张忠德等[14]报道7 例UCD患儿,5 例为浆细胞型,2 例MCD 患儿浆细胞型和混合型各1 例。
本文5 例UCD 患儿均表现为淋巴结进行性增大,直径4.0 ~7.0 cm。其中3 例累及颈部淋巴结,1 例为纵隔淋巴结,1 例为腹膜后淋巴结,颈部肿大淋巴结发生率较高,与成人好发于纵隔、腹部的特点有所不同。患者病初多无明显临床症状,随着淋巴结逐渐增大,可产生局部压迫症状,根据压迫部位不同而产生不同的症状。本文例2 病程36个月,颈部淋巴结增大后出现气道压迫症状,颈部CT 平扫显示左侧颈部淋巴结增大,气管压迫变窄并向右侧移位,增强CT 则显示淋巴结明显强化。
本文例4 为UCD 合并PNP,以皮肤黏膜的多形性皮疹起病,临床表现复杂,肿大淋巴结位于腹膜后,给临床诊断带来一定的困难。PNP 是1990 年由Anhalt 等[15]首先描述的一种自身免疫性皮肤病,表现为皮肤黏膜的多形性皮疹,其特点是易并发良性和恶性肿瘤,其中伴发CD 者约占10%。
4 例MCD 除多处淋巴结肿大外,还伴有发热、盗汗、乏力、消瘦和肝脾肿大等全身症状,实验室检查有贫血、ESR加快、丙种球蛋白增多等改变,与文献报道相似。另外,本文有2 例病史较长的患儿出现生长发育迟缓,可能与该病炎性因子表达过多,患儿处于长期发热、疾病消耗状态,影响正常生长发育有关。例8 于疾病缓解后出现身高、体重追赶性增长,例9 因干骺端已闭合,随诊身高无明显改善。
CD 临床表现复杂,若临床医生缺乏对CD 的认识,很难早期发现并诊断,出现漏诊和误诊。尤其是MCD 患儿表现为多部位淋巴结肿大并伴有多系统功能紊乱,实验室检查多存在异常,临床表现呈现多样化,又无特异性,可靠的诊断需要经过淋巴结切除术,通过典型的病理形态及免疫组化确立诊断。文献中行淋巴结穿刺活检者病理报告几乎均为“淋巴组织增生”或“慢性淋巴结炎”,故穿刺活检对确诊本病意义不大。
按照CD 的诊断标准,除了具备特征性的病理改变,诊断很大程度上还依赖于排除其他原发性疾病,因此,临床上应注意排除感染性疾病( 结核感染、慢性活动性EB 病毒感染、HIV 感染、寄生虫感染) ,结缔组织病( 系统性红斑狼疮、幼年型特发性关节炎、白塞病、过敏性紫癜、干燥综合征等) ,肿瘤性疾病( 淋巴瘤、胸腺瘤、浆细胞瘤、神经鞘瘤、转移癌等) 。
由于CD 存在明显异质性,目前尚无统一和标准的治疗方案。治疗包括早期手术切除、化疗、放疗、抗病毒以及靶向生物制剂等[16]。UCD 患儿治疗以手术切除为主,预后良好,绝大多数可长期存活,极少有复发者。本组5 例UCD患儿接受手术切除后,4 例无合并症患儿均达到临床缓解,随访3 年11 月至8 年10 月无复发。1 例合并PNP 者接受联合治疗,病情无明显改善。Nikolskaia 等[17]总结分析了28 例成人CD 合并PNP 患者,手术及免疫抑制治疗效果不理想,其中22 例最终死于呼吸衰竭,提示CD 合并PNP 者预后不良。MCD 需要全身治疗,手术切除受累淋巴结可以改善部分患者的临床症状,但疗效短暂。有学者认为,少数没有全身症状的患者可随诊观察,不需要积极干预。但因MCD 大多具有侵袭性,大部分患者需要联合化疗。本组4例MCD 患儿中,1 例行标准化CHOP 方案化疗后达临床缓解,其余3 例MCD 患儿经手术切除肿大淋巴结( 其中1 例脾切除) 后,临床表现逐渐缓解,实验室检查指标改善,因家属顾虑未行化疗,目前维持临床缓解16 ~36 个月。Parez等[12]报道MCD 患儿的预后要明显地好于成人,可能与起病年龄小的儿童疾病进展较成人慢、侵袭性较弱有关。近年来,根据对CD 发病机制的研究,出现一些新的靶向治疗药物,如抑制IL-6 自分泌的蛋白酶抑制剂、抗CD20 的单克隆抗体等[18,19],为CD 的治疗提供了良好的前景。
[1]Frizzera G. Castleman's disease and related disorders. Semin Diagn Pathol,1988,5:346-364
[2]张之南,主编.血液病诊断及疗效标准.第3 版.北京:北京科学出版社.2007:228-229
[3]Wu XY(吴晓燕), Song HM, Shen J,et al.Castleman disease with paraneoplastic pemphigus in a child.Chin J Pediatr(中华儿科杂志),2008,46(2): 149-150
[4]Castleman B, Towne VW. Case records of the Massachusetts General Hospital: case 32-1984. N Engl J Med,1954,311:388-398
[5]Castleman B, Iverson L, Menendez VP. Localized mediastinal lymph node hyperplasia resembling thymoma.Cancer,1956,9:822-830
[6]Han X(韩潇), Zhou DB.Advances in etiology and management of Castleman's disease.Acta Academiae Medicinae Sinicae(中国医学科学院学报),2009,31(5):639-643
[7]Brandt S,Bodine D,Dunbar C. Dysregulated interleukin 6 expression produces a syndrome resembling Castleman's disease in mice.J Clin Invest,1990,86(2):592-599
[8]Dupin N, Diss TL, Kellam P, et al. HHV-8 is associated with a plasmablastic variant of Castleman disease that is linked to HHV-8-positive plasmablastic lymphoma. Blood,2000,95(4):1406-1412
[9]Amin HM, Medeiros LJ, Manning JT, Jones D.Dissolution of the lymphoid follicle is a feature of the HHV8 + variant of plasma cell Castleman's disease.Am J Surg Pathol,2003,27(1):91-100
[10]Talat N, Belgaumkar AP, Schulte KM. Surgery in Castleman's disease: a systematic review of 404 published cases. Ann Surg,2012,255(4):677-684
[11]Huang S(黄爽), Zhou CJ, Jin M, et al.Clinical characteristics of 5 children with Castleman's disease and review of literature.Chin J Pediatr(中华儿科杂志),2010,48(8): 625-628
[12]Parez N, Bader-Meunier B, Roy CC,et al.Paediatric Castleman disease: report of seven cases and review of the literature.Eur J Pediatr,1999,158(8): 631-637
[13]Smir BN, Greiner TC, Weisenburger DD. Multicentric lymph node hyperplasia in children: a clinicopathologic study of eight patients. Mod Pathol,1996,9(12):1135-1142
[14]Zhang ZD(张忠德), Xi ZJ, Wu XR, et al. Castleman disease in 9 cases and literature review.J Appl Clin Pediatr(实用儿科临床杂志),2005,20(5):450-451
[15]Ardudt GJ,Kim SC,Stanley JR,et a1.Paraneoplastic pemphigus.An autoirnmune mueocutaneous disease associated with neoplasia.N Engl J Med,1990,323(25):1729-1735
[16]Saeed-Abdul-Rahman I, Al-Amri AM.Castleman disease.Korean J Hematol,2012,47(3):163-177
[17]Nikolskaia OV,Nousari CH,Anhalt GJ.Paraneoplastic pemphigus in association with Castleman's disease. Br J Dermatol,2003,149: 1143-1151
[18]Ocio E,Sanchez-Guijo F,Diez-Campelo M.Efficacy of rituximab in an aggressive form of multicentric Castleman disease associated with immune phenomena.Am J Hematol,2005,78(4):302-305
[19]Hess G, Wagner V, Kreft A, et al.Effects of bortezomib on pro-inflammatory cytokine levels and transfusion dependency in a patient with multicentric Castleman disease.Br J Haematol,2006,134(5):544-545
猜你喜欢
中国临床医学(2022年3期)2022-07-08
临床超声医学杂志(2022年4期)2022-05-07
临床与实验病理学杂志(2021年7期)2021-09-06
天津医科大学学报(2021年4期)2021-08-21
天津医科大学学报(2021年3期)2021-07-21
医学信息(2021年5期)2021-03-21
放射学实践(2020年5期)2020-06-01
养生保健指南(2019年11期)2019-12-17
中国临床医学影像杂志(2018年2期)2018-08-02
中国医学影像技术(2018年5期)2018-05-18
