
腓骨肌萎缩症临床表现、基因分型和分子发病机制研究进展
2013-03-11 08:19:52晖综述宋福聪审校
中风与神经疾病杂志 2013年10期
郭 鹏,翟 晖综述,宋福聪审校
腓骨肌萎缩症(Charcot-Marie-Tooth disease,CMT)是一组最常见的具有高度临床和遗传异质性的遗传性周围神经病,不同人群中CMT 的发病率略有不同,约在17/100,000~40/100,000 之间[1]。自1991 年发现由17 号染色体短臂11.2区(17p11.2)1.5Mb 的正向串联重复突变导致CMT1A,1992 年PMP22 基因被克隆以来的二十年内,目前已有39 个CMT 基因位点被定位,28 个疾病基因被克隆(http://www.ncbi.nlm.nih.gov/OMIM/)。遗传方式可为常染色体显性遗传、常染色体隐性遗传和X 连锁遗传。常染色体显性遗传CMT 依据神经病理学改变可分为CMT1(脱髓鞘型)和CMT2(轴索型),常染色体隐性遗传CMT 被命名为CMT4,X 连锁遗传的CMT 被命名为CMTX[2],近几年又发现一些中间型常染色体显性遗传CMT 病例,既有神经脱髓鞘,又有神经元轴突变性,被命名为DI-CMT[3],各型又根据相关基因及特异临床表现再分为若干亚型。本文就CMT 的临床表现、基因分型和分子发病机制研究进展进行综述。
1 CMT 的临床表现
CMT 患者多在10 岁前发病,少数患者在成年发病,病程进展缓慢。CMT 的临床表现具有一些相似之处,常表现为进行性对称性肢体远端肌无力和肌萎缩,部分患者可伴远端感觉减退或缺失、骨骼畸形、腱反射减弱或缺失。肌萎缩常由下肢开始逐渐发展到上肢,大腿下1/3 以下肌肉无力和萎缩,形成“鹤腿”或倒置酒瓶样畸形,行走和跑步困难,跨阈步态。手部骨间肌和大小鱼际肌无力和萎缩,出现爪形手或猿手畸形,肌萎缩一般不超过肘关节以上,手的精细动作不能。四肢近端肌肉萎缩较为少见,仅出现在一些症状较重的患者中[4]。下肢比上肢更易出现末梢型感觉障碍,通常痛温觉和振动觉均减退,位置觉较少受损,也是先累及足部,再向小腿延伸,然后累及手部。腱反射减弱或缺失,可伴自主神经功能障碍和营养障碍。常伴弓形足、脊柱侧弯等骨骼畸形。少数患者可先出现扁平足,然后转变为弓形足。晚期发病的CMT 患者往往无足部骨骼畸形。其他常见的症状和体征包括肌肉痛性痉挛、双足发冷、发绀和过度角质化等。发病极早的病例可导致肌张力低下,运动发育迟缓,踮脚走路。
除上述CMT 共同的临床特征外,一些类型CMT 可出现特有的神经受累症状。如CMT1D 可出现脑神经受损,CMT1X 可出现中枢神经受损,CMT2A 可出现视神经萎缩,CMT2C 可出现声带麻痹及呼吸受累,CMT2D 上肢受累严重,CMT2J 可出现Adie’s 瞳孔,CMT4B2 可出现早发性青光眼,CMT4C 可出现重度脊柱侧弯,CMT4F 感觉缺失明显,HMNSR可出现听力丧失,DI-CMT B 可出现神经痛。但不是所有该型CMT 患者均有这些特征症状。在同一CMT 家系中,不同个体出现的临床症状轻重有所不一,有的个体出现典型的CMT 症状,而有的个体仅有轻微症状,甚至有的携带致病基因个体无症状。
根据周围神经电生理和病理特点可将CMT 分为两型:CMT1(脱髓鞘型),其正中神经运动传导速度<38m/s,神经活检示广泛的节段性脱髓鞘和髓鞘增生形成洋葱球样结构;CMT2(轴索型),其正中神经运动传导速度正常或轻度减慢(>38m/s),神经活检示轴索变性。区别与经典的CMT 临床分型,中间型CMT 作为一组正中神经传导速度介于25~45m/s,神经病理兼具脱髓鞘和轴索变性特点的CMT 变异型正逐步被认识[5]。
2 CMT 的基因分型
CMT 的遗传方式以常染色体显性遗传最多见,见于大部分CMT1 和CMT2 家系患者,其次为X 连锁显性遗传;常染色体隐性遗传较少见,而散发病例并不少见。依据CMT 的遗传位点和疾病基因可将CMT 分为CMT1、CMT2、CMT4、CMTX 和DI-CMT5 类(见表1)。90% 以上的CMT 是 由PMP22、GJB1、MPZ 和MFN2 这4 种基因突变引起的。由17p11.2 区包含PMP22 基因在内的1.5Mb 的正向串联重复突变所致的CMT1A 约占CMT 患者总数的40%~50%,GJB1基因突变所致的CMTX1 约占CMT 总数的7%~12%,MFN2基因突变导致CMT2A 是CMT2 最常见的基因型,GDAP1 基因突变是CMT 常染色体隐性遗传最常见的基因型。同一疾病基因突变可引起不同类型的CMT,如MPZ 基因突变可引起CMT1B、CMT2I、CMT2J,GDAP1 基因突变可引起CMT2H、CMT2K 和CMT4A,EGR2 基因突变可引起 CMT1D 和CMT4E。不同疾病基因突变也可引起相同类型CMT,如MFN2 和KIF1B 突变引起CMT2A,MTMR2 和SBF2 基因突变均可导致CMT4B。
3 CMT 的分子发病机制
3.1 髓鞘功能异常 周围神经是雪旺氏细胞、神经元轴索和间质细胞相互作用的整体,它们之间的信号转导是周围神经的正常发育和维持结构完善和损失修复必不可少的,任何细胞成分的基因缺陷或其他损伤都将导致其他细胞成分的结构和功能缺陷,从而出现周围神经的功能障碍。PMP22 为主要在雪旺氏细胞表达的跨膜蛋白,除作为髓鞘结构蛋白外,尚参与调节雪旺氏细胞的增殖、分化和凋亡,过度表达的PMP22 不能进行正常的细胞内转移而积聚在Golgi复合体中,影响雪旺氏细胞的正常增生和分化;也可能由于PMP22 与髓鞘蛋白零(myelin protein zero,MPZ)组成一个复合体以保持髓鞘的稳定,在CMT1 患者中PMP22/MPZ 的比例升高,破坏了髓鞘的稳定性[6]。MPZ 编码周围神经髓磷脂的主要结构蛋白即髓鞘蛋白0 蛋白,突变可影响MPZ 蛋白的所有成分,导致髓磷脂附着减少以及突变蛋白分布异常[7]。影响黏附功能的突变可导致严重早期发病的神经病,而影响信号传导功能的突变则与较轻的晚发神经病相关。而EGR2是雪旺氏细胞的转录调控因子,它结合在DNA 的特定区域调节基因的活动,可以作用于与构成髓鞘相关的几个基因调节PMP22、MPZ 和CX32 等髓鞘蛋白的合成[8]。CX32 的周围神经表达位于郎飞结周和Schmidt 2 Lanterman 切迹处,相邻细胞膜上CX32 六聚体之间形成的间隙连接通道在雪旺细胞近轴索和远轴索的胞浆之间信息传递和物质交换中起重要作用。Preiaxin 特异表达于形成髓鞘的雪旺细胞,由于mRNA 的不同剪切形成L2Preiaxin 和S2Preiaxin,其中S2Preiaxin 弥散分布于胞质,而L2Preiaxin 位于靠基底膜侧的雪旺细胞膜,与肌萎缩相关蛋白2(dystrophin 2 related protein 2)和营养不良聚糖蛋白(dystroglycan)形成复合物,从而连接雪旺细胞的细胞骨架和基底膜,其生物学功能可能为稳定
成熟的周围神经髓鞘[9]。
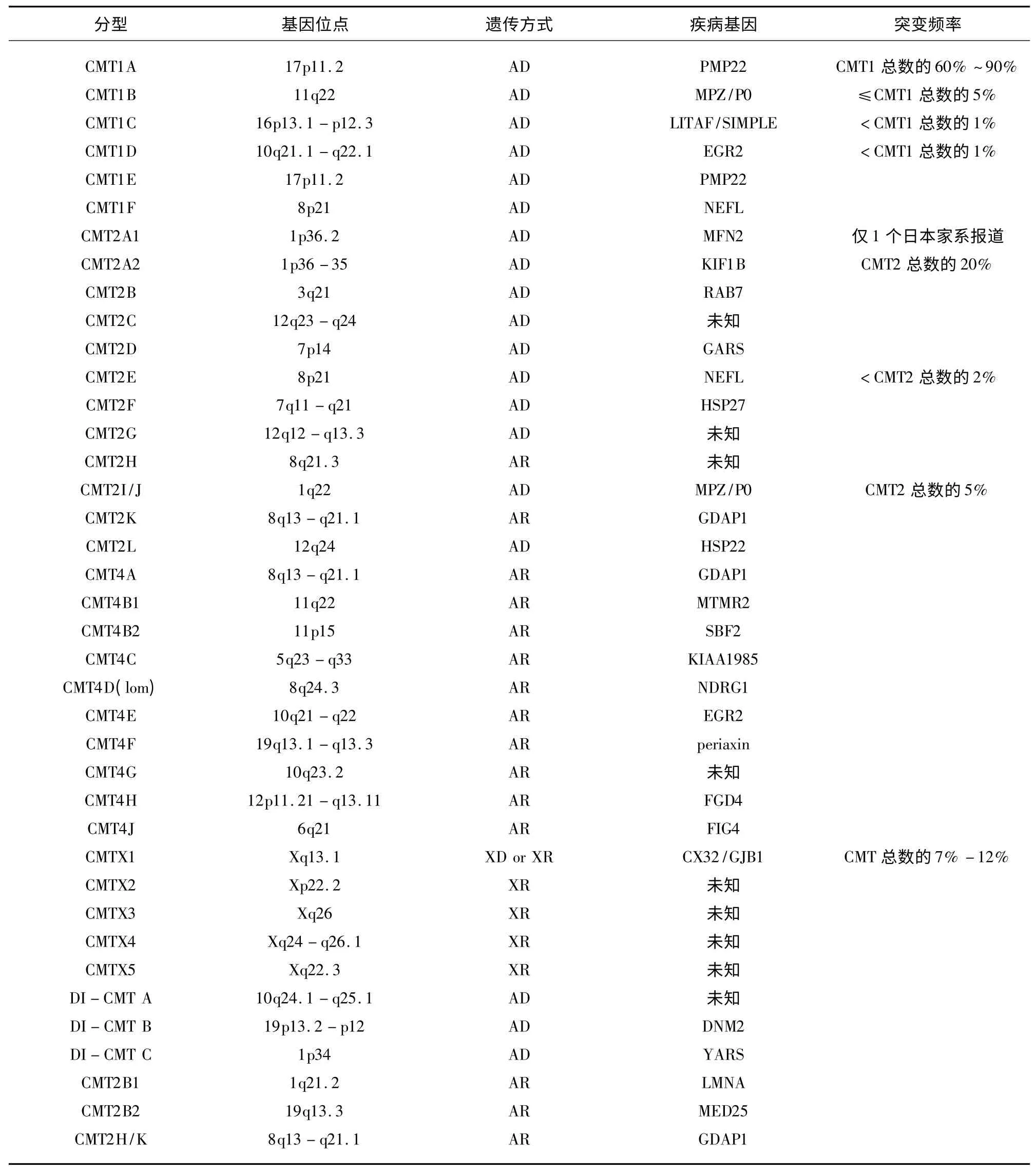
表1 CMT 基因型及疾病基因一览表
3.2 轴索运输和蛋白质转运功能缺陷 KIF1B 编码驱动蛋白,属于运动蛋白家族成员,在轴索运输中起重要作用[10]。突变蛋白与微管结合后的ATP 酶不能被激活或激活后活性明显降低,轴突的快速运输受到限制,所需的营养物质和结构蛋白供给受限,从而导致轴突变性引起CMT2A1 临床表型。MFN2 基因的突变多数位于其功能区,改变了MFN2蛋白的空间结构,破坏了其调节线粒体动态平衡的功能,引起线粒体网络结构的破坏和能量代谢障碍[11]。轴突不能合成物质,所需的营养物质和结构蛋白需由胞体提供,MFN2 基因突变导致的能量代谢障碍限制了轴突营养物质的获得,从而引起轴突变性,临床上表现为CMT2A2 表型。
SIMPLE 编码的蛋白位于膜周溶酶体,可能为E3 泛素连接酶通过泛素介导的蛋白质降解途径,参与蛋白质和其他物质的降解。尽管SIMPLE 基因在体内很多种组织和细胞中表达,但其突变后仅引起脱髓鞘性神经病,提示可能跟PMP22基因突变一样由于雪旺细胞中特定的蛋白降解受阻所[12]。RAB7 编码的蛋白位于细胞内间隙,在囊泡运输和细胞内吞通路中起重要作用。RAB7 蛋白调节从细胞表面到溶酶体的转运,是维持溶酶体正常功能所必需的[13]。
3.3 小分子热休克蛋白相关的退行变 小热休克蛋白(small heat shock p roteins,sHSPs)的共同结构特点为均含有一个α-晶体蛋白区,该区是位于蛋白C 端一段含85~100 个氨基酸的高度保守序列。到目前为止共发现10 种小热休克蛋白。HSP22 和HSP27 基因突变导致遗传性周围神经病的发现为CMT 发病机制研究提供了新思路。sHSPs 的生物学功能包括参与细胞内信号转导、抗凋亡、稳定细胞骨架蛋白和分子伴侣功能[14]。研究证实,突变Hsp27 蛋白可降低神经元细胞的生存能力,它可以破坏神经丝蛋白的组装,并进一步形成细胞内蛋白聚集物,神经丝为轴突细胞骨架的主要成分,对轴突直径和轴突转运起关键作用,Evgrafov 等[15]认为Hsp22 与Hsp27 存在相互作用,Hsp22 参与Hsp27 蛋白活性的调节,Hsp27 蛋白突变后引起轴突细胞骨架的崩溃和轴突转运障碍,最终导致轴突变性、死亡而致病。
3.4 其他机制 CMT2D 与编码甘氨酸tRNA 合成酶的GARS 基因突变相关,该酶在蛋白质合成过程中负责把甘氨酸加入到蛋白质氨基酸链适当位置。神经细胞的正常功能需要含有丰富甘氨酸的蛋白质,而这些蛋白质的活性都可能由于GARS 基因的突变而降低。Antonellis 等[16]研究证实,GARS 基因的突变降低了甘氨酰tRNA 合成酶的活性,使甘氨酸合成受阻,从而导致了神经冲动传导受阻或富含甘氨酸的神经元受损,这可能是GARS 基因突变导致CMT2D 表型的发病机制。
综上所述,CMT 的诊断依据的是临床、电生理和病理资料,但是由于CMT 具有明显的临床异质性,不同基因突变所致的CMT 患者从表型上很难区分,而且并非所有患者都有典型的临床或电生理改变,另外,神经活检的损伤比较大,患者接受程度低。相比之下基因诊断的准确性高,无损伤,并且在病程早期即可做出诊断确定基因型。
[1]Lee YC,Yu CT,Lin KP,et al.MPZ mutation G123S characterization evidence for a complex pathogenesis in CMT disease[J].Neurology,2008,70(4):273-277.
[2]Pennuto M,Tinelli E,Malaguti M,et al.Ablation of the UPRmediator CHOP restores motor function and reduces demyelination in Charcot-Marie-Tooth 1B mice[J].Neuron,2008,57(3):393-405.
[3]Nicholson G,Myers S.Intermediate forms of Charcot-Marie-Tooth neuropathy.A review[J].Neuromolecular Med,2006,8(1):123-130.
[4]Kabzinska D,Hausmanowa-Petrusewicz I,Ochanski A.Charcot marie tooth disorders with an autosomal recessive mode of inheritance[J].Clin Neuropathol,2008,27(1):1-12.
[5]Barisic N,Claeys KG,Sirotovic-Skerlev M,et al.Charcot Marie Tooth disease:a clinico-genetic confrontation[J].Ann Hum Genet,2008,72(3):416-441.
[6]Nobbio L,Vigo T,Abbruzzese M,et al.Impairment of PMP22 transgenic Schwann cells differentiation in culture:implications for Charcot-Marie-Tooth type 1A disease[J].Neurobiol Dis,2004,16(1):263-273.
[7]Runker AE,Kobsar I,Fink T,et al.Pathology of a mouse mutation in peripheral myelin protein P0 is characteristic of a severe and early onset form of human Charcot-Marie-Tooth type 1B disorder[J].Cell Biol,2004,165(4):565-573.
[8]Berger P,Young P,Suter U,et al.Molecular cell biology of Charcot-Marie-Tooth disease[J].Neurogenetics,2002,4(1):12-15.
[9]Guilbot A,Williams A,Ravise N,et al.A mutation in periaxin is responsible for CMT4 F,an autosomal recessive form of Charcot2 Marie 2 Tooth disease[J].Hum Mol Genet,2001,10(4):415-421.
[10]Zhao C,Takita J,Tanaka Y,et al.Charcot-Marie-Tooth disease type 2A caused by mutation in a microtubule motor KIF1Bbeta[J].Cell,2001,105(5):587-597.
[11]Kijima K,Numakura C,Izumino H,et al.Mitochondrial GTpase mitofusin 2 mutation in Charcot-marie-tooth neuropathy type 2A[J].Hum Genet,2005,116(1):23-27.
[12]Bennett CL,Shirk AJ,Huynh HM,et al.SIMPLE mutation in demyelinating neuropathy and distribution in sciatic nerve[J].Ann Neurol,2004,55(5):713-720.
[13]Verhoeven K,Jonghe P,Coen K,et al.Mutations in the small GTPase late endosomal protein RAB7 cause Charcot-Marie-Tooth type 2B neuropathy[J].Am Hum Genet,2003,72(3):722-727.
[14]Kappe G,Franck E,Verschuure P,et al.The human genome encodes 10 alpha 2 crystallin 2 related small heat shock p roteins:HspB 1-10[J].Cell Stress Chaperones,2003,8(1):53-61.
[15]Evgrafov OV,Mersiyanova I,Irobi J,et al.Mutant small heat-shock protein 27 causes axonal Charcot-Marie-Tooth disease and distal hereditary motor neuropathy[J].Nat Genet,2004,36(6):602-606.
[16]Antonellis A,Ellsworth RE.Glycyl tRNA synthetase mutations in charcot-marie-tooth disease type 2D and distal spinal muscular atrophy type[J].Am J Hum Genet,2003,72(5):1293-1299.
猜你喜欢
中华耳科学杂志(2022年1期)2022-11-24 15:09:22
生物化学与生物物理进展(2022年11期)2022-03-02 11:34:48
浙江临床医学(2021年12期)2021-11-29 14:43:18
健康之家(2021年19期)2021-05-23 09:10:44
党的生活(黑龙江)(2018年9期)2018-10-17 01:24:24
益寿宝典(2018年1期)2018-01-27 01:50:24
中国卫生标准管理(2015年4期)2016-01-14 05:16:45
山东医药(2015年16期)2016-01-12 00:40:07
吉林大学学报(医学版)(2015年1期)2015-12-17 07:47:27
中华神经创伤外科电子杂志(2015年1期)2015-01-21 09:09:24
