
咪唑-[1,2-b]哒嗪类mTOR 抑制剂的合成及抗肿瘤活性研究
2013-03-08 08:36翁怡然高善云杨振军张亮仁张礼和
中国药物化学杂志 2013年6期
翁怡然,高善云,杨振军,张亮仁 ,张礼和
(北京大学 医学部 天然药物及仿生药物国家重点实验室,北京100191)
恶性肿瘤因其难于早期发现、诊断和治疗已成为严重威胁人类健康的常见病。作用于细胞内信号通路的关键靶点的小分子抑制剂是一类新型的抗肿瘤药物。磷酸肌醇3-激酶PI3K/人雷帕霉素靶蛋白mTOR 信号通路成为近年来抗肿瘤药物研究的热点。
人雷帕霉素靶蛋白mTOR(mammalian target of rapamycin)是哺乳动物丝氨酸/苏氨酸蛋白激酶,由2549 个氨基酸组成,分子量289 kDa,属PI3K 系列激酶PIKK 家族[1]。mTOR 在细胞内以两种复合物形式存在,分别是mTORC1 和mTORC2[2]。mTORC1 的主要功能是调节蛋白质合成和细胞周期进程,而mTORC2 则在肌动蛋白细胞骨架组织和细胞存活方面发挥重要作用[3-4]。目前第一代mTOR 抑制剂雷帕霉素及其类似物,如坦西莫司(temsirolimus)、依维莫司(everolimus,RAD001)、ridaforolimus 分 别 于2007、2009、2005 年经FDA 批准用于肾癌和骨肉瘤的治疗[5-6],使得mTOR 成为被临床证实的有效抗癌药物靶点。但由于雷帕霉素只能作用在mTORC1,对mTORC2 作用不强,且临床上仅对部分肿瘤有效(如肾癌、骨肉瘤),因此选择性的mTORC1/mTORC2 双重抑制剂,即ATP 竞争性mTOR 抑制剂成为该信号通路的研究热点[7-9]。目前小分子mTOR 抑制剂有AZD-2014、INK-128 处于临床Ⅰ期研究,OSI-027 处于临床Ⅱ期阶段[6]。

Figure 1 The structure of WYE-687 and WYE-354
Yu 等[10-14]设计合成了一系列吡唑并嘧啶类mTOR 抑制剂(图1),如化合物WYE-687(IC50=0.007 μmol·L-1)、WYE-354(IC50=0.005 μmol·L-1)。该类化合物的构效关系研究表明:1)吗啉及其衍生物可显著提高吡唑并嘧啶类化合物与mTOR的结合能力,极大地提升了该类mTOR 抑制剂的活性和选择性;2)在6 位芳基上引入脲类及酰胺类取代基团可大幅提升化合物的活性,IC50可达到亚纳摩尔级别;3)在6 位为芳基脲类取代的同时,在1 位引入哌啶衍生物,可显著提高化合物对mTOR 的选择性抑制作用。
基于对mTOR 结构及其与抑制剂结合特征的分析,本研究利用骨架迁越及生物电子等排原理,并以化合物WYE-354 为模型化合物,设计了咪唑-[1,2-b]哒嗪类mTOR 抑制剂。目标化合物结构均经1H-NMR、13C-NMR 及HRMS 确证。合成路线见图2。
目标化合物的合成共分5 步。第一步是母核的构建,将3-氨基-4-溴-6-氯哒嗪、1-(N-Boc-哌啶-4-基)溴乙醛在加热条件下回流,得到咪唑并哒嗪母核;第二步是通过碱性条件下的亲核取代反应,在母核8 位引入吗啉类结构;第三步脱去哌啶N 原子上的Boc 保护基,连接所设计的一系列基团;第四步是通过Suzuki 偶联反应在母核6 位引入苯胺结构;最后一步是在碱性条件下与相应的胺类化合物进行三光气反应,得到一系列终产物。

Figure 2 The synthetic route to target compounds 13 -28
1 合成实验
化合物的核磁共振数据由Bruker AVANCE III 400 MHz 核磁共振波谱仪测定,由MestReNova(Ver.6.1.0,Mestrelab Research S. L. )软件处理;质谱数据(ESI-TOF)由QSTAR 液质联用仪测定;高分辨质谱数据(ESI-TOF)由Bruker Apex IV FTMS 型傅立叶离子回旋变换质谱仪测定;熔点由毛细管法测定,温度未经校正;薄层色谱硅胶板及柱色谱硅胶(200 ~300 目,300 ~400 目)购自上海上邦实业有限公司。所有溶剂、原料和试剂如无说明均为市售分析纯。
1.1 8-溴-6-氯-3-(N-叔丁氧羰基-哌啶-4-基)-咪唑并[1,2-b]哒嗪(1)的合成
将3-氨基-4-溴-6-氯哒嗪0.5 g(2.4 mmol)和1-(N-叔丁氧羰基-哌啶-4-基)溴乙醛12 mmol 加入到5 mL 无水乙醇中,加热回流24 h。过滤干燥得褐色固体0.64 g,置于装有30 mL 乙醇的烧瓶中,加入碳酸钾0.45 g、二碳酸二叔丁酯(Boc2O)0.7 g,室温反应12 h。过滤,柱色谱分离得白色固体0.55 g,收率55%,mp 100 ~102 ℃。
1H-NMR(400 MHz,CDCl3)δ:7.60(s,1H),7.34(s,1H),4.24(s,2H),3.29(s,1H),2.94(s,2H),2.07(d,J=21.4 Hz,2H),1.83 ~1.60(m,2H),1.49(s,9H)。MS(ESI-TOF+)m/z:415.3[M +H]+。
1.2 8-(4-吗啉基)-6-氯-3-(N-叔丁氧羰基-哌啶基)-咪唑并[1,2-b]哒嗪(2)的合成
将化合物1 0.58 g(1 mmol)溶解于9 mLN,N-二甲基甲酰胺中,滴加吗啉0.13 mL(1.1 mmol)、三乙胺0.23 mL(1.1 mmol),回流24 h。过滤,柱色谱分离得淡黄色固体0.5 g,收率81%,mp 130 ~132 ℃。1H-NMR(400 MHz,CDCl3)δ:7.29(s,1H),6.06(s,1H),4.23(s,2H),4.14 ~3.65(m,8H),3.27(s,1H),2.94(s,2H),2.20 ~2.01(m,2H),1.64(d,J=11.6 Hz,2H),1.53(d,J=34.2 Hz,9H)。MS(ESI-TOF+)m/z:422.3[M +H]+。
1.3 (S)-8-(3-甲基吗啉基-4-基)-6-氯-3-(N-叔丁氧羰基-哌啶基)-咪唑并[1,2-b]哒嗪(3)的合成
方法同化合物2,只是将原料吗啉改为(S)-3-甲基 吗 啉,收 率75%,mp 140 ~142 ℃。1H-NMR(400 MHz,CDCl3)δ:7.28(s,1H),6.02(s,1H),5.22(s,1H),4.45(s,1H),4.27(dd,J=23.6,6.5 Hz,2H),4.13 ~4.00(m,1H),3.90(d,J=11.5 Hz,1H),3.86 ~3.67(m,2H),3.50(td,J=12.5,3.5 Hz,1H),3.27(t,J=11.8 Hz,1H),2.94(t,J=11.5 Hz,2H),2.12(d,J=12.6 Hz,2H),1.76 ~1.57(m,2H),1.49(s,9H),1.36(d,J=6.7 Hz,3H)。MS(ESI-TOF+)m/z:436.4[M +H]+。
1.4 3-(N-甲氧羰基-哌啶-4-基)-8-(4-吗啉基)-6-氯-咪唑并[1,2-b]哒嗪(4)的合成
将化合物2 0.6 g(1.42 mmol)溶于3.5 mL二氯甲烷溶液中,冰浴下滴加三氟乙酸1.2 mL,室温反应12 h。加2 mol·L-1碳酸钠水溶液中和,二氯甲烷萃取,无水硫酸钠干燥,旋干,得油状物0.52 g。将上述油状物加入到装有8 mL 二氯甲烷的烧瓶中,冰浴下滴加三乙胺(0.45 mL,3.2 mmol)和氯甲酸甲酯(0.16 mL,1.9 mmol),室温反应12 h。饱和碳酸氢钠溶液洗涤,二氯甲烷萃取,无水硫酸钠干燥,柱色谱分离[V(石油醚)∶V(乙酸乙酯)=2∶1]得黄色固体0.42 g,收率65%,mp 132 ~134 ℃。1H-NMR(400 MHz,CDCl3)δ:7.29(s,1H),6.08(s,1H),4.25(dd,J=14.3,8.6 Hz,2H),4.09 ~3.97(m,4H),3.97 ~3.84(m,4H),3.75(d,J=10.6 Hz,3H),3.41 ~3.18(m,1H),3.01(t,J=12.3 Hz,2H),2.14(d,J=12.3 Hz,2H),1.78 ~1.62 (m,2H)。MS(ESI-TOF+)m/z:380.4[M +H]+。
1.5 (S)-3-(N-甲氧羰基-哌啶-4-基)-8-(3-甲基吗啉基-4-基)-6-氯-咪唑并[1,2-b]哒嗪(5)的合成
方法同化合物4,只是将原料化合物2 改为化合物3、氯甲酸甲酯改为甲磺酰氯,收率63%,mp 136 ~138 ℃。1H-NMR(400 MHz,CDCl3)δ:7.29(s,1H),6.03(s,1H),5.23(s,1H),4.60 ~4.17(m,3H),4.12 ~4.00(m,1H),3.91(dd,J=11.5,2.8 Hz,1H),3.86 ~3.65(m,5H),3.51(td,J=12.6,3.8 Hz,1H),3.30(ddd,J=11.9,7.7,3.5 Hz,1H),3.00(d,J=11.5 Hz,2H),2.15(d,J=12.3 Hz,2H),1.67(s,2H),1.37(d,J=6.8 Hz,3H)。MS(ESI-TOF+)m/z:394.4[M +H]+。
1.6 3-(N-甲磺酰基-哌啶-4-基)-8-(4-吗啉基)-6-氯-咪唑并[1,2-b]哒嗪(6)的合成
方法同化合物4,只是将原料氯甲酸甲酯改为甲磺酰氯,收率68%,mp 96 ~98 ℃。1H-NMR(400 MHz,CDCl3)δ:7.32(d,J=7.3 Hz,1H),6.09(d,J=3.2 Hz,1H),4.27 ~3.82(m,10H),3.24(dd,J=9.5,6.0 Hz,1H),3.03 ~2.76(m,5H),2.27(d,J= 11.3 Hz,2H),1.90(td,J=12.7,3.9 Hz,2H)。MS(ESI-TOF+)m/z:400.3[M +H]+。
1.7 (S)-3-(N-甲磺酰基-哌啶-4 基)-8-(3-甲基吗啉基-4 基)-6-氯-咪唑并[1,2-b]哒嗪(7)的合成
方法同化合物4,只是将原料化合物2 改为化合物3、氯甲酸甲酯改为甲磺酰氯,收率70%,
mp 98 ~100 ℃。1H-NMR(400 MHz,CDCl3)δ:7.32(s,1H),6.04(s,1H),5.26(s,1H),4.49(s,1H),4.07(dd,J=11.7,3.6 Hz,1H),3.92(dd,J= 20.9,8.8 Hz,3H),3.79 (dd,J= 27.6,7.2 Hz,2H),3.52(d,J=3.7 Hz,1H),3.25(s,1H),2.99 ~2.87(m,2H),2.85(s,3H),2.27(d,J=12.6 Hz,2H),1.97 ~1.80(m,2H),1.37(d,J=6.8 Hz,3H)。MS(ESI-TOF+)m/z:414.4[M +H]+。
1.8 3-(N-甲氧羰基-哌啶-4-基)-8-(4-吗啉基)-6-(2-氨基苯基-5-基)-咪唑并[1,2-b]哒嗪(8)的合成
将化合物4 0.2 g(0.53 mmol)、4-氨基苯硼酸频哪醇酯0.1 g(0.76 mmol)、四三苯基膦钯0.1 g(0.1 mmol)加入到11 mLN,N-二甲基甲酰胺中,加入2 mol·L-1碳酸钠水溶液0.8 mL,氩气保护,120 ℃下反应12 h。水洗,二氯甲烷萃取,无水硫酸钠干燥,硅胶柱色谱分离[V(二氯甲烷)∶V(甲醇)=150∶1],得淡黄色固体0.17 g(含杂质)。
1.9 (S)-3-(N-甲氧羰基-哌啶-4-基)-8-(3-甲基吗啉基-4-基)-6-(2-氨基苯基-5-基)-咪唑并[1,2-b]哒嗪(9)的合成
方法同化合物8,只是将原料化合物4 改为化合物5,收率72%,mp 126 ~128 ℃。1H-NMR(400 MHz,CDCl3)δ:7.71(d,J=8.0 Hz,2H),7.31(s,1H),6.77(d,J=7.6 Hz,2H),6.37(s,1H),3.88(dt,J=62.6,12.7 Hz,7H),3.53(t,J=12.5 Hz,1H),3.34(t,J=11.9 Hz,1H),3.03 ~2.77(m,6H),2.35(d,J=11.0 Hz,2H),2.02 ~1.88(m,2H),1.32(d,J=6.6 Hz,3H)。MS(ESITOF+)m/z:451.4[M +H]+。
1.10 3-(N-甲磺酰基-哌啶-4-基)-8-(4-吗啉基)-6-(2-氨基苯基-5-基)-咪唑并[1,2-b]哒嗪(10)的合成
方法同化合物8,只是将原料化合物4 改为化合物6,产物含杂质。
1.11 (S)-3-(N-甲磺酰基-哌啶-4-基)-8-(3-甲基吗啉基-4-基)-6-(2-氨基苯基-5-基)-咪唑并[1,2-b]哒嗪(11)的合成
方法同化合物8,只是将原料化合物4 改为化合物7,收率70%,mp 118 ~120 ℃。1H-NMR(400 MHz,CDCl3)δ:7.71(d,J=8.0 Hz,2H),7.31(s,1H),6.77(d,J=7.6 Hz,2H),6.37(s,1H),3.88(dt,J=62.6,12.7 Hz,7H),3.53(t,J=12.5 Hz,1H),3.34(t,J=11.9 Hz,1H),3.03 ~2.77(m,6H),2.35(d,J=11.0 Hz,2H),2.02 ~1.88(m,2H),1.32(d,J=6.6 Hz,3H)。MS(ESITOF+)m/z:471.4[M +H]+。
1.12 3-(N-叔丁氧羰基-哌啶-4-基)-8-(4-吗啉基)-6-(2-氨基苯基-5-基)-咪唑并[1,2-b]哒嗪(12)的合成
方法同化合物8,只是将原料化合物4 改为化合物2,产物含杂质。
1.13 目标化合物13 ~28 的合成通法
将化合物8 ~12(0.1 mmol)加入二氯甲烷1 mL 中,滴 加 三 乙 胺57 μL,加 入 三 光 气(0.12 mmol),室温搅拌10 min。将上述混合液滴加至相应胺类(0.6 mmol)的二氯甲烷(1 mL)溶液中,室温反应12 h。水洗,二氯甲烷萃取,干燥,硅胶柱色谱分离[V(二氯甲烷)∶V(甲醇)=50∶1],得到目标化合物13 ~28。目标化合物的结构及理化及波谱数据见表1。

Table 1 Physical constants and spectra data of target compounds

Continued Table 1
2 抗肿瘤活性评价
2.1 目标化合物与mTOR 的对接
mTOR 结构由PI3Kγ(3IBE)同源模建得到,去除受体中的配体小分子,对蛋白加氢,赋CHARMm 力场,作为分子对接的受体。配体小分子赋力场。使用Gold 3.0.3 进行分子对接实验,设定10 个对接构象,选取打分数值最高的结合模式,用PyMOL v 0.99 分析对接结果。
图3 为化合物16 与mTOR 对接示意图,对接结果证明,化合物16 能够进入mTOR 的ATP竞争性作用口袋,并且吗啉环上的氧原子与Val2240 有氢键作用,芳基脲的氧原子分别与Asp2198、Phe2358 形成氢键作用。
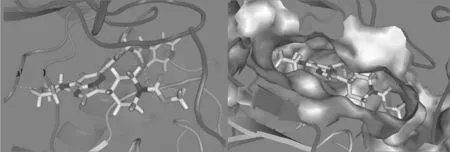
Figure 3 The schematic of compound 16 combined with mTOR active sites
2.2 对人癌细胞株的体外增殖抑制实验
选取对mTOR 高表达的肿瘤细胞株:人肺癌细胞株A549、H460、人黑色素瘤细胞株A375、人乳腺癌细胞株MCF-7、人脑胶质瘤细胞株U87,采用SRB 法,以AZD8055 为阳性对照药,测试化合物的体外抗细胞增殖活性。设定给药时间为72 h,每个测试组至少设定3 个复孔,重复两次实验得平均IC50值。实验结果见表2。
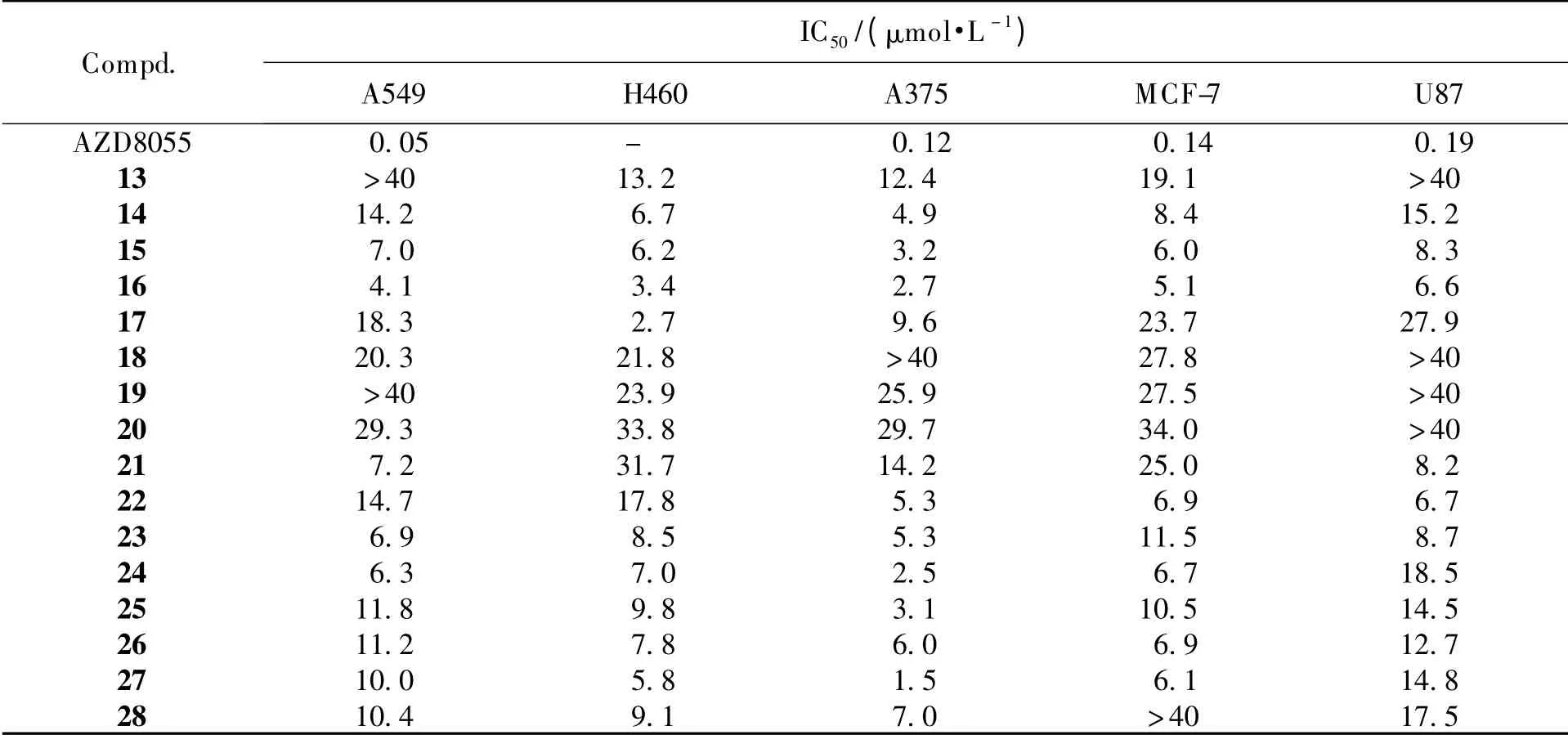
Table 2 The antitumor activity of target compounds in vitro
2.3 mTOR 激酶抑制实验
mTOR 激酶抑制实验由Invitrogen 公司完成,使用Z'-LYTE(r)测试方法。选取活性化合物16、27(对A549、H460、A375、MCF-7、U87 细胞增殖的平均IC50值为4.3 μmol·L-1和7.6 μmol·L-1)进行mTOR 激酶抑制实验,初筛设定在10 μmol·L-1和1 μmol·L-1两种浓度,结果见表3。
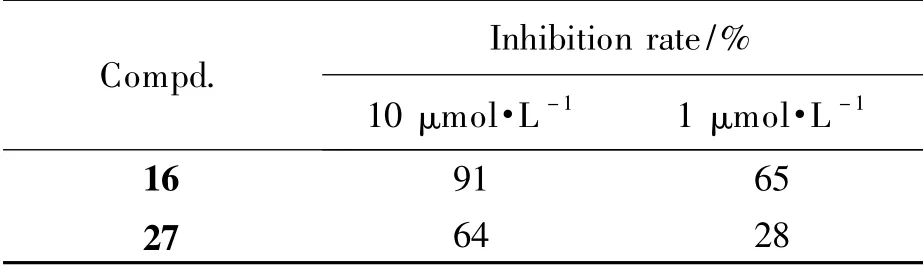
Table 3 The inhibition rate of compounds 16,27 against mTOR enzymatic activity
对于1 μmol·L-1的抑制率大于50% 的化合物16 进行IC50值的测试,结果表明,化合物16 对mTOR 激酶有一定的抑制作用,IC50为274 nmol·L-1。
3 讨论
3.1 化学合成
化合物1 的合成分为两步,第一步是关环反应,所得产物的哌啶环N 原子上的Boc 保护基被脱除,故需在第二步重新连接Boc 基。第一步的产物为盐酸盐,不溶于反应溶剂乙醇,作者起初采用的后处理方法是用饱和碳酸钠溶液洗涤、二氯甲烷萃取、干燥、柱色谱分离,操作繁琐。后对反应完成后析出的褐色固体进行结构鉴定,发现其中杂质含量很少,可适当减少溶剂用量,使产物充分析出,过滤干燥后直接进行下一步反应,操作更为简便。
化合物8 的后处理,作者起初采用反应完成后直接旋干溶剂、柱色谱分离的方法,后发现下一步反应收率总是偏低,副产物较多,分析其原因,很可能是由于化合物8 中混有反应溶剂DMF,影响了其与三光气的反应。因此,在反应后需先用水洗去DMF,再进行柱色谱分离。
3.2 活性测试
体外细胞增殖实验结果表明,化合物能够对A549、H460、A375、MCF-7 和U87 肿瘤细胞有不同程度的抑制作用,基于体外抗肿瘤活性结果发现:1)化合物的R3为吡啶基对提高活性有利;2)8 位为S-甲基吗啉基的化合物活性优于吗啉基取代的化合物,尤其是对肺癌细胞的抑制作用有明显提高;3)R2为甲磺酰基时,因化合物溶解性下降,活性降低。mTOR 激酶抑制实验表明,化合物16 对mTOR 有较强的抑制作用(IC50=247 nmol·L-1)。以上实验结果,将作为进一步结构改造的依据。
[1] 潘智,张令强,蒋继志,等.mTOR 的研究进展[J].细胞生物学杂志,2006,28(3):395 -398.
[2] LAPLANTEL M,SABATINI D.mTOR signaling in growth control and disease[J].Cell,2012,149(2):274 -293.
[3] CORNU M,ALBERT V,HALL M N.mTOR in aging,metabolism,and cancer[J]. Curr Opin Genet Dev,2013,23(1):53 -62.
[4] JEWELL J L,GUAN K L. Nutrient signaling to mTOR and cell growth[J]. Trends Biochem Sci,2013,38(5):233 -242.
[5] FASOLO A,SESSA C.mTOR inhibitors in the treatment of cancer[J]. Expert Opin Investig Drugs,2008,17(11):1717 -1734.
[6] ZAYTSEVA Y Y,VALENTINO J D,GULHATI P,et al.mTOR inhibitors in cancer therapy[J].Cancer Lett,2012,319(1):1 -7.
[7] MARTINS F,OLIVEIRA M A,WANG Q,et al. A review of oral toxicity associated with mTOR inhibitor therapy in cancer patients[J].Oral Oncol,2013,49(4):293 -298.
[8] GARCíA-ECHEVERRíA C. Allosteric and ATPcompetitive kinase inhibitors of mTOR for cancer treatment[J]. Bioorg Med Chem Lett,2010,20(15):4308 -4312.
[9] YU K,SHI C,TORAL-BARZA L,et al.Beyond rapalog therapy:preclinical pharmacology and antitumor activity of WYE-125132,an ATP-competitive and specific inhibitor of mTORC1 and mTORC2[J].Cancer Res,2010,70(2):621 -631.
[10] YU K,TORAL-BARZA L,SHI C,et al. Biochemical,cellular,andin vivoactivity of novel ATP-competitive and selective inhibitors of the mammalian target of rapa-mycin[J].Cancer Res,2009,69(15):6232 -6240.
[11] YU K,ZASK A,VERHEIJEN J C,et al. ATP-competitive inhibitors of the mammalian target of rapamycin:design and synthesis of highly potent and selective pyrazolopyrimidines[J].J Med Chem,2009,52(16):5013 -5016.
[12] YU K,NOWAK P,COLE D C,et al. Discovery of potent and selective inhibitors of the mamma-lian target of rapamycin(mTOR)kinase[J].J Med Chem,2009,52(22):7081 -7089.
[13] YU K,VERHEIJEN J C,RICHARD D J,et al.Discovery of 4-morpholino-6-aryl-1H-pyrazolo[3,4-d]pyrimidines as highly potent and selective ATP-competitive inhibitors of the mammalian target of rapamycin(mTOR):optimization of the 6-aryl substituent[J].J Med Chem,2009,52(24):8010 -8024.
[14] YU K,ZASK A,KAPLAN J,et al. Morpholine derivatives greatly enhance the selectivity of mammalian target of rapamycin(mTOR)inhibitors[J]. J Med Chem,2009,52(24):7942 -7945.Abstract:The mammalian target of rapamycin(mTOR),a central regulator of growth,survival,and metabolism,is a validated target for cancer therapy.We have designed a series of imidazo[1,2-b]pyridazines and explored the anticancer activities and structure-activity relationships.Starting form 3-amino-4-bromo-6-chloropyridazine and bromo-1-(N-Boc-piperidin-4-yl)acetaldehydeviathe creation of imidazo[1,2-b]pyridazine and Suzuki coupling reaction,totally 16 compounds were synthesized and all of them were not reported yet,and their structures were confirmed by HRMS,1H-NMR and13C-NMR. All of the compounds showed diverse inhibitory activity of cellular proliferation(cell lines A549,H460,A375,MCF-7 and U87),among which the compounds 15,16 and 27 have potent activity in cellular proliferation assays.The structureactivity relationships reveals that the compounds with pyridyl on the urea have strongest inhibitory activity,and replacement of morpholino to(S)-3-methylmorpholino improves the inhibitory activity.
猜你喜欢
健康体检与管理(2022年2期)2022-04-15
广州化工(2022年3期)2022-02-24
农药科学与管理(2019年8期)2019-11-23
农药科学与管理(2019年12期)2019-05-20
上海化工(2018年10期)2018-10-31
合成化学(2015年1期)2016-01-17
应用化工(2014年7期)2014-08-09
中国药理学通报(2014年2期)2014-05-09
郑州大学学报(理学版)(2014年4期)2014-03-01
化工生产与技术(2014年5期)2014-02-27
