
原发性侧索硬化2例报道及相关文献复习
2013-01-31 02:15王凤志何志义邓淑敏赵奕楠
中风与神经疾病杂志 2013年3期
王凤志,何志义,李 蕾,邓淑敏,刘 芳,赵奕楠,孟 肃
原发性侧索硬化2例报道及相关文献复习
王凤志,何志义,李 蕾,邓淑敏,刘 芳,赵奕楠,孟 肃
目的 探讨原发性侧索硬化(PLS)的临床诊断、与经典的肌萎缩侧索硬化的关系、神经电生理特点、影像学特点及鉴别诊断。方法 分析我科收治的2例PLS患者的临床资料,并复习相关文献。结果 PLS是隐匿起病,进展缓慢的仅累及上运动神经元的神经退行性疾病,很多初诊为PLS的患者经过长期随访最后发展为肌萎缩侧索硬化,PLS的影像学表现多样,神经电生理检查容易早期发现下运动神经元损伤。结论 对于疑诊PLS的患者,明确是否具有局限性下运动神经元损伤症状十分重要,对患者进行时间的纵断随访对疾病最后的诊断和预后评估意义重大。
原发性侧索硬化; 肌萎缩侧索硬化; 上运动神经元; 下运动神经元
原发性侧索硬化(primary lateral sclerosis,PLS)最早由Erb提出[1],是罕见、特发性、非家族遗传性的仅累及上运动神经元(uppor motor neuron,UMN)的神经退行性疾病[2]。大约占运动神经元病(motor neuron disease,MND)的1%~3%。国内仅有少数报道。本文通过分析我科收治的2例原发性侧索硬化患者的临床资料,并结合相关文献对其临床诊断、与经典的肌萎缩侧索硬化症(amyotrophic lateral sclerosis,ALS)的关系、神经电生理特点、影像学特点及鉴别诊断等进行探讨。
1 临床资料
例1,女,57岁,以“走路易摔跤4年半,蹲起费力半年”为主诉入院。患者于4年半前无明显诱因出现走路易摔跤,自感双下肢僵硬,不能控制自己脚步,前脚跟踩后脚尖,易向前摔倒,偶尔感觉下背部肌肉僵硬。近半年出现蹲起费力。病来无饮水呛咳及吞咽困难。既往体健,否认高血压、冠心病、糖尿病病史,家族成员无类似疾病史。查体:神清语利,发育正常,心肺腹正常,脑神经正常,痉挛步态,四肢肌力Ⅴ级,无肌萎缩及肌束颤动,双下肢折刀样肌张力增高,BCR、TCR(⧺,⧺),PSR、ASR(⧻,⧻),Hoffmann征(-,-),Babinski征(+,+)。双侧指鼻试验及跟膝胫试验正常,深浅感觉正常。化验检查:血常规、尿常规、血沉、风湿三项、CTDⅢ、肝肾功能、血电解质、血糖、血脂、凝血三项、心肌酶谱、甲功系列、甲状腺抗体系列、乙肝、人免疫缺陷病毒抗体、梅毒螺旋体特异抗体均正常,ANA:1∶40。MMSE评分26分,其中计算功能减3分,执行功能减1分,患者未上过学。肌电图显示:左右正中神经、右尺神经感觉神经传导速度减慢,诱发电位波幅降低,左尺神经感觉神经传导速度减慢。颈段、胸段及腰骶段磁共振显示:C4-5,T11-12间盘膨出,L3-4,L5-S1间盘膨出,脊髓无受压,髓内未见异常信号。头部MRI显示双侧顶叶皮质下-内囊后肢可见对称性斑片状略长T2信号,FLAIR序列呈高信号(见图1、图2)。
例2,男,41岁,以“双下肢无力1年半”为主诉入院。患者于1年半前受凉后出现双下肢无力,左下肢为重,走路拖地,2~3min之后好转,恢复至正常。之后上述症状偶有出现,均在情绪紧张、激动及气温变化时出现,持续2~3min能恢复至正常。2011年12月份开始病情逐渐加重,表现为下楼费力,抬腿费力,走路迈不开步,左下肢不能完成踢腿动作,不能跑、跳。既往体健,否认高血压、糖尿病、心脏病史。否认家族遗传病史。查体:神清语利,发育正常,心肺腹正常,脑神经正常,痉挛步态,四肢肌力Ⅴ级,无肌萎缩及肌束颤动,四肢肌张力正常。BCR、TCR、PSR、ASR(⧻,⧻),Hoffmann征(+,+),Babinski征(+,+),踝阵挛(+,+)。针刺痛觉、深感觉未见确切异常。轮替试验及指鼻试验双侧稳准,跟膝胫试验双侧稳准,Romberg征阴性。化验检查:血常规、尿常规、肝肾功能、血电解质、血糖、血脂、凝血三项、心肌酶谱、乙肝、贫血系列、人免疫缺陷病毒抗体、梅毒螺旋体特异抗体均正常。颈椎MRI+增强示颈椎间盘退变,颈3-7间盘突出,脊髓无受压变形。胸部正侧位未见异常改变。肌电图示左腓总神经运动神经诱发电位波幅降低,传导速度正常。头部MRI显示中央前回大脑皮质局限性萎缩(见图3)。
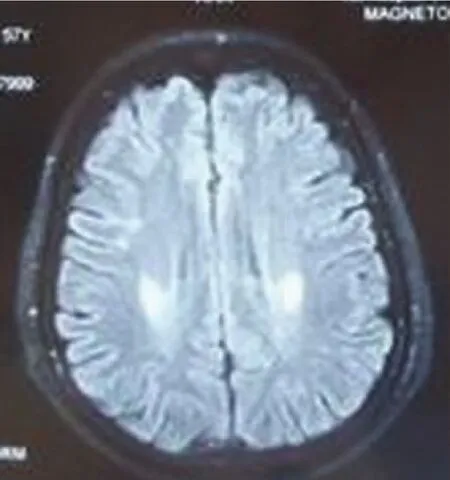
图1 Flair序列示双侧侧脑室旁放射冠区对称性斑片状高信号
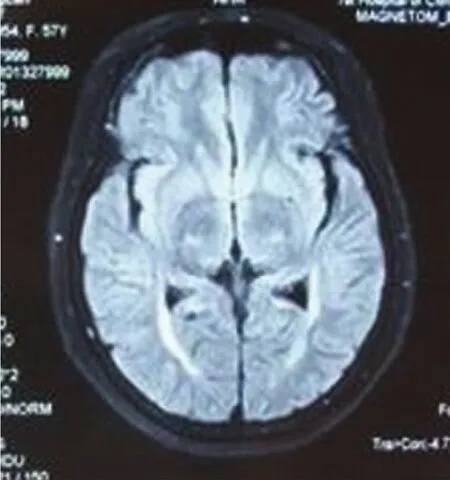
图2 Flair序列示双侧内囊后肢锥体束走行区对称性斑片状高信号
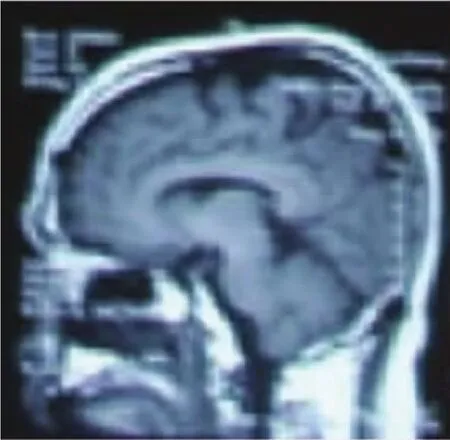
图3 T1序列示中央前回大脑皮质局限性萎缩
2 讨论
PLS是MND的一种类型,MND是一组病因未明的选择性侵犯脊髓前角细胞、脑干后组运动神经元、皮质锥体细胞及锥体束的慢性进行性神经变性病,包括肌萎缩侧索硬化(ALS)、原发性侧索硬化(PLS)、进行性肌萎缩(progressive muscle atrophy,PMA)和进行性延髓麻痹(progressive bulbar palsy,PBP)。
2.1 诊断
1992年,Pringle等[3]基于对8例患者的临床特点分析和1例尸检提出了PLS的临床诊断标准: (1)隐袭起病,通常从下肢开始;(2)无此类症状的家族史;(3)对称性,逐渐进展的病程;(4)成年起病,起病年龄通常在50岁以后;(5)病程大于3年且未出现临床表现或肌电图明显提示的下运动神经元(lower motor neuron,LMN)受损症状;(6)累及皮质脊髓束,伴或不伴皮质脑干束功能障碍;(7)影像学和实验室检查结果除外其他疾病。但随着有关PLS的陆续报道,发现Pringle等的诊断标准具有临床不确定性,因为其中的一些患者最终有LMN症状出现,并且诊断标准中没有区分那些单纯具备UMN症状和同时存在轻微LMN受损改变的患者[4]。针对特定症状出现多长时间才能诊断PLS,也是众口不一,因为随着时间的延长,初始诊断为PLS的患者很可能发展成为UMN主导的ALS(UMN-D ALS)甚至ALS[5]。Pringle及Tartaglia等[2]认为至少3年未发现LMN受损征象才能诊断为PLS,而Gordon等[6]通过随访发现PLS患者平均3.17年出现肌电图的异常,之后又6m,即初诊PLS 3.77年后出现LMN受损症状。故认为从发生症状开始4年后,是诊断PLS较可靠的时间。本文两例患者均缓慢隐袭起病,首发症状为双下肢无力、痉挛步态,查体腱反射亢进,病理征阳性,无家族史,所有症状和体征均限于锥体束损害,无感觉障碍及肌萎缩征象。例1发病时间超过3年,根据Pringle提出的标准,可临床诊断为PLS。例2发病时间较短,但临床也仅表现出UMN受损征象,PLS可能性大。
2.2 PLS与ALS的关系
PLS与ALS是两个独立的疾病还是运动神经元变性连续过程中的两个不同阶段,这个问题一直备受争议。Tan等[7]对一位82岁PLS患者尸检发现UMN系统,包括中央前回、下行锥体束的严重变性。但也发现少量LMN内出现ALS的典型改变,即胞浆包涵体。证明在PLS中LMN同时受累,但神经元的变性速度很慢,故认为PLS是罕见的以UMN受累为主的ALS的一种形式。Forestier等[8]对9例PLS患者前瞻性,纵断面的研究表明PLS不仅仅累及UMN,LMN同样受累,其与ALS的区别在于LMN受累的程度和病变稳定性。虽然ALS和PLS有许多相似之处,甚至说PLS是ALS的一种形式,但两者也有明显的差别。Hudson等[9]和Forestier一项对20例PLS患者进行的研究[1]发现PLS的病程明显较ALS长,且不出现明显的肌萎缩和肌力减弱,假性球麻痹进展较ALS缓慢。Tartaglia等[2]回顾性比较研究661例ALS患者及43例PLS患者,通过首发症状及随访中出现的不同临床表现来鉴别两种疾病。研究发现:肢体僵硬是PLS(47%)和ALS (4%)唯一明显不同的主诉症状;另外,8%ALS患者以吞咽困难为主诉,而PLS中没有患者以此为主诉。随访中发现肢体瘫痪在PLS(2%)中的出现率明显低于ALS(100%),在43例PLS患者中,其中1人随访24年,16人随访大于10年,均未出现肢体无力所致的瘫痪;同时发现PLS的平均生存期明显长于ALS。皮质症状在PLS患者中略多见,但临床上显著的痴呆在ALS中较常见。故认为以肢体僵直为主诉就诊且3年内不产生肢体瘫痪的患者患PLS的可能性大。Gordon等[6]对9例 PLS、15例UMN-D ALS及10例典型的ALS患者的初发症状及随访进行研究,发现PLS患者初诊时肌力明显高于UMN-D ALS,UMN-D ALS组高于典型ALS组。与ALS组相比,PLS组症状初发部位更多见于下肢,就诊时症状存在时间明显长于另两组,从侧面反映了病程进展的缓慢性。与PLS组相比,UMN-D ALS组和ALS组更易发生明显的肌萎缩、体重减轻及吞咽困难。本文两例患者就诊时肌力均Ⅴ级,无肌萎缩、肌束颤动及球麻痹症状,病来体重无明显减轻,病例1肢体僵硬显著,与文献研究一致。
2.3 神经电生理及头磁共振表现
2.3.1 神经电生理表现 Weber等[10]研究了12例PLS及12例ALS患者的皮质运动神经元传导,发现PLS患者疾病进展缓慢,上运动神经元受损症状与经颅皮质磁刺激高阈值及中枢性运动传导时间延长有关。而ALS患者经颅皮质磁刺激阈值在疾病早期即降低,且中枢性运动传导时间通常正常,说明PLS与ALS皮质运动神经元的损伤程度不同。Upmeijer等[11]研究了10例PLS患者的神经电生理表现,发现PLS是进展缓慢的,以UMN受损为突出表现的疾病,但肌电图也显示了轻度的LMN受损症状,如混合型最大自主收缩电位和运动单位诱发电位(MUP)的延长。本文两例患者肌电图表现不特异,不支持LMN受损,尚有待于长期随访复查。
2.3.2 头磁共振表现 Forestier等[1]观察了20例PLS的患者:MRI正常者6例,轻度弥漫性皮质萎缩8例,双侧颞底部皮质萎缩2例,轻度白质疏松3例,轻度初级运动区皮质萎缩1例。没有患者显示典型的皮质脊髓束走行区高信号。Upmeijer等[11]研究10例PLS患者的MRI表现,显示中央前回皮质萎缩,萎缩可延伸到顶枕叶区域,中央前回平均表面积约减少到正常的75%,颅脑矢状位时显示明显,这种局部的皮质萎缩较ALS更常见。10位患者中仅有1位显示锥体束走行区内囊后肢T2高信号。Charles等[12]报道了1例PLS患者8.5年内3次颅脑MRI的动态变化。发现就诊时、就诊后4.7年、就诊后8.5年的头MR显示中央前回皮质萎缩进行性加重,8.5年时,顶上区、运动前区、额前区也明显萎缩,故认为PLS时受累的皮质虽然有局限性,但要比以往尸检报道的PLS神经元的丢失局限于运动皮质锥体细胞广泛得多。本文病例1患者头MR显示较典型的锥体束走行区长T2信号,病例2示中央前回大脑皮质局限性萎缩,符合PLS的影像学表现。
2.4 鉴别诊断
典型的PLS表现为UMN受累引起的痉挛状态,故需与下列疾病相鉴别:(1)多发性硬化(multiple sclerosis,MS):两者病灶虽然都以脑白质为主,但临床上MS多为急性或亚急性起病,而PLS隐袭起病;MS病程中有缓解与复发,PLS则为进行性发展;MS常有感觉障碍,PLS则无此现象;MRI所见MS病灶大多分布于脑室周围区,病灶长轴与脑室垂直,PLS病灶则局限于上运动神经元分布区。(2)额颞叶痴呆:两者都可出现认知功能障碍,故临床需鉴别。Mochizuki等[13]曾报道1例以上运动神经元症状为主诉,后出现帕金森症状和行为异常,伴有典型的中央前回,新纹状体及额颞叶变性及神经元泛素包涵体形成的额颞叶痴呆病例,证明额颞叶痴呆早期也可出现上运动神经元受损症状。Richard等[14]对9例诊断为PLS的患者进行神经心理学测试,发现9例患者不存在痴呆,但是其中8例患者都存在轻度认知功能障碍。但额颞叶痴呆一般早期出现人格改变,情感变化及言语障碍可以鉴别。此外,PLS还应与进行性多灶性白质脑病、神经梅毒、脊髓亚急性联合变性,遗传性痉挛性截瘫、颈椎病等疾病加以鉴别,通过相关病史、血清学检查、明确家族史及颈椎磁共振不难鉴别。
2.5 治疗与预后
PLS作为神经变性疾病之一,目前尚无治疗方法。Zhai等[15]通过对25例PLS患者的临床表现及神经电生理进行分析,发现其中56%的患者具有相似的临床进展过程,即“上行性”进展,即患者的痉挛状态首先出现在下肢,疾病缓慢稳定的进展,平均3.6年后出现上肢的痉挛状态,后又平均1.5年,出现球症状,包括语言障碍等。这种临床表现相似的亚组的出现也许有利于进一步探讨疾病的病因及治疗。对于疑诊PLS的患者,明确是否具有局限性下运动神经元损伤症状十分重要。只具备UMN受损的PLS患者临床预后较好。所以对病程在4年内,疑诊PLS的患者进行时间的纵断随访对疾病最后的诊断和预后评估尤为重要。文中所诉两例患者均需经过长期随访以明确诊断。
[1]Le Forestier N,Maisonobe T,Piquard A,et al.Does primary lateral sclerosis exist?A study of 20 patients and a review of the literature[J].Brain,2001,124(10):1989-999.
[2]Tartaglia MC,Rowe A,Findlater K,et al.Differentiation between primary lateral sclerosis and amyotrophic lateral sclerosis:Examination of symptoms and signs at disease onset and during follow-up[J].Arch Neurol,2007,64(2):232-236.
[3]Pringle CE,Hudson AJ,Munoz DG,et al.Primary lateral sclerosis. Clinical features,neuropathology and diagnostic criteria[J].Brain,1992,115:495-520.
[4]Gordon PH,Cheng B,Katz IB,et al.The natural history of primary lateral sclerosis[J].Neurology,2006,66:647-653.
[5]Bruyn RP,Koelman JH,Troost D,et al.Motor neuron disease(amyotrophic lateral sclerosis)arising from longstanding primary lateral sclerosis[J].J Neurol Neurosurg Psychiatry,1995,58(6):742-744.
[6]Gordon PH,Cheng B,Katz IB,et al.Clinical features that distinguish PLS,upper motor neuron-dominant ALS,and typical ALS[J].Neurology,2009,72(22):1948-1952.
[7]Tan CF,Kakita A,Piao YS,et al.Primary lateral sclerosis:a rare upper-motor-predominant form of amyotrophic lateral sclerosis often accompanied by frontotemporal lobar degeneration with ubiquitinated neuronal inclusions?Report of an autopsy case and a review of the literature[J].Acta Neuropathol,2003,105(6):615-620.
[8]Le Forestier N,Maisonobe T,Spelle L,et al.Primary lateral sclerosis:further clarification[J].Journal of the Neurological Sciences,2001,185:95-100.
[9]Hudson AJ,Kiernan JA,Munoz DG,et al.Clinicaopathological features of primary lateral sclerosis are different from amyotrophic lateral sclerosis[J].Brain Res Bull,1993,30(34):359-364.
[10]Weber M,Stewart H,Hirota N,et al.Corticomotoneuronal connections in primary lateral sclerosis(PLS)[J].Amyotroph Lateral Scler Other Motor Neuron Disord,2002,3(4):190-198.
[11]Kuipers-Upmeijer J,de Jager AEJ,Hew JM,et al.Primary lateral sclerosis:clinical,neurophysiological,and magnetic resonance findings[J].J Neurol Neurosurg Psychiatry,2001,71(5):615-620.
[12]Smith CD.Serial MRI findings in a case of primary lateral sclerosis[J].Neurology,2002,58(4):647-649.
[13]Mochizuki A,Komatsuzaki Y,Iwamoto H,et al.Frontotemporal dementia with ubiquitinated neuronal inclusions presenting with primary lateral sclerosis and parkinsonism:clinicopathological report of an autopsy case[J].Acta Neuropathol,2004,107(4):377-380.
[14]Caselli RJ,Smith BE,Osborne D,et al.Primary lateral sclerosis:a neuropsychological study[J].Neurology,1995,45(11):2005-2009.
[15]Zhai P,Pagan F,Statland J,et al.Primary lateral sclerosis:A heterogeneous disorder composed of different subtypes[J]?Neurology,2003,60(8):1258-1265.
Report of two cases of primary lateral sclerosis and review of the literature
WANG Feng-zhi,HE Zhi-yi,LI Lei,et al.(Department of Neurology,The First Affiliated Hospital of China Medical University,Shenyang110001,China)
ObjectiveTo investigate the clinical diagnosis of primary lateral sclerosis(PLS),and the relationship with the classic amyotrophic lateral sclerosis(ALS),the neural electrophysiological features,imaging features and differential diagnosis.MethodWe analyzed the clinical data of 2 patients with PLS who were admitted to our hospital,and reviewed of relevant literature.ResultsPLS is a neurodegenerative disease characterized by involving only the upper motor neuron,occult onset,and the slow progress.Mmany patients diagnosed as PLS after long-term follow-up turned out to be ALS,PLS is various in imaging manifestation,and neural electrophysiological examination is helpful in early detection of lower motor neuron injury.ConclusionFor patients suspected with PLS,identifying the existence of localized injury of lower motor neuron symptoms is very important,conducting a longitudinal follow-up is of great significance to the last diagnosis and prognosis.
Primary lateral sclerosis;Amyotrophic lateral sclerosis;Uppor motor neuron;Lower motor neuron
R744
A
1003-2754(2013)01-0244-04
2012-11-01;
2012-12-25
(中国医科大学附属第一医院神经内科,辽宁 沈阳110001)
通迅作者:何志义,E-mail:hezhiyi0301@sina.com
文章编号:1003-2754(2013)01-0248-03
猜你喜欢
保健与生活(2022年13期)2022-07-06
体育科技文献通报(2021年12期)2021-12-18
疯狂英语·新阅版(2021年8期)2021-09-10
世界科学技术-中医药现代化(2021年10期)2021-03-02
小资CHIC!ELEGANCE(2021年46期)2021-01-11
睿士(2020年11期)2020-11-16
考试与评价·高二版(2020年2期)2020-09-10
VOGUE服饰与美容(2020年9期)2020-09-02
北京广播电视报(2019年8期)2019-03-27
武警医学(2019年2期)2019-03-05
